

 English
English文献解读|Mol Cancer(37.3):DNMT1 靶向重塑整体 DNA 低甲基化,以增强口腔鳞状细胞癌的肿瘤抑制并避免毒性
✦ +
+
论文ID
原名:DNMT1-targeting remodeling global DNA hypomethylation for enhanced tumor suppression and circumvented toxicity in oral squamous cell carcinoma
译名:DNMT1 靶向重塑整体 DNA 低甲基化,以增强口腔鳞状细胞癌的肿瘤抑制并避免毒性
期刊:Molecular Cancer
影响因子:37.3
发表时间:2024.05.16
DOI号:10.1186/s12943-024-01993-1
背 景
DNA 甲基化稳态的忠实维持离不开 DNA 甲基转移酶 1 (DNMT1) 在癌症进展中的作用。目前已确定 DNMT1 是口腔鳞状细胞癌 (OSCC) 的潜在候选靶点。然而,如何利用 DNMT1 相关的整体 DNA 甲基化来调节 OSCC 仍不清楚。
实验设计
结 果
01

DNMT1 过表达在口腔肿瘤转化过程中逐渐增加,并与 OSCC 中的肿瘤生长有关
为了评估 DNMT1 在 OSCC 发展过程中的表达,研究团队检测了从口腔正常组织到增生和发育不良病变最终到 OSCC 组织的人类样本。结果显示,随着口腔肿瘤转化的过程,DNMT1 表达逐渐增加,在 OSCC 组织中达到峰值(图 1 A)。利用了一系列 OSCC 细胞系,最初证实其 DNMT1 表达明显高于 NOK 细胞。然后使用代表性 Cal27 和 FaDu 细胞生成 DNMT1 敲低细胞系(sh-DNMT1-1 和 sh-DNMT1-2)。鉴于 sh-DNMT1-1 的基因沉默效果更明显,因此他们选择它进行后续研究。 DNMT1 沉默导致细胞增殖显著减少,表现为体外 BrdU 和 Ki67 表达下降以及 OSCC 细胞球形成能力下降(图 1B-C)。此外,他们还建立了 OSCC 异种移植小鼠模型(图 1 D)。sh-DNMT1 转染的癌细胞表现出显著降低的致瘤能力,表现为肿瘤生长减少、细胞增殖减少和细胞凋亡增加(图 1E-G)。
利用 GSE30784 数据集的转录组分析(RNA-seq)数据(图S2A-C),他们观察到口腔肿瘤转化过程中 DNMT1 表达逐渐增加(图 1 H)。随后,从 TCGA 数据库中获取了原发性 OSCC 和正常组织,并对具有匹配的临床病理和随访记录的更大的患者队列进行了进一步分析。与邻近的正常组织相比,DNMT1 在 OSCC 组织中过表达(图 1 I)。使用患者的临床病理和随访数据进行平滑曲线拟合显示,DNMT1 表达与 OSCC 死亡风险之间存在非线性和动态关联,尽管差异并不显著(图 1 J)。简而言之,在感染点 2.92 之前,随着 DNMT1 表达的增加,死亡风险呈总体上升趋势;此后,潜在关系逆转。除了多元 Cox 回归或 Kaplan ‒ Meier 分析等线性分析(图S1 E-F)之外,他们还选择使用限制性立方样条分析来评估它们的非线性关系。该分析表明,DNMT1 表达与总体生存风险比之间存在波动关系(图 1 K)。值得注意的是,癌症中 DNMT1 表达的显著下降与死亡率和总体生存风险比的显著降低相关。泛癌分析证实了 DNMT1 在包括头颈癌在内的各种癌症类型中过表达(图S2 D),间接表明 DNMT1 是一个潜在靶点。这些究结果表明,DNMT1 过表达有助于口腔恶性转化的启动并促进癌细胞的恶性行为,导致 OSCC 肿瘤的生长。
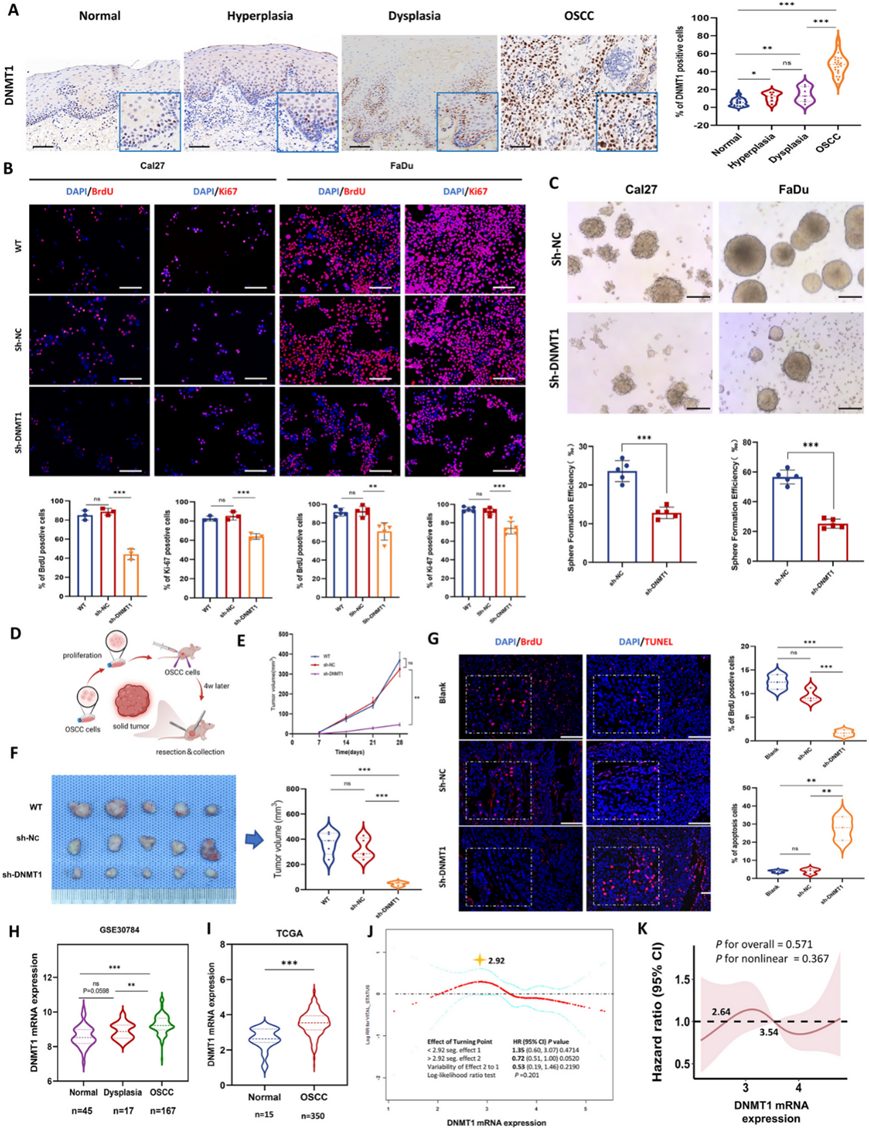
图1. DNMT1 表达随口腔肿瘤转化而增加,其过表达与肿瘤生长相关。
(a) 代表性 IHC 图像和分析。(b) OSCC细胞系中的 BrdU 和 Ki67 的免疫荧光。(c) OSCC细胞系中的 BrdU 和 Ki67 的免疫荧光。(d) 显示异种移植 OSCC 小鼠模型的示意图。(e) 肿瘤生长曲线。(f) 研究结束时 OSCC 异种移植肿瘤的展示和肿瘤体积统计数据。(g) OSCC异种移植肿瘤中的 BrdU 和 TUNEL 免疫荧光和分析。(h) DNMT1在癌变的多个阶段中的 mRNA 表达。(i) DNMT1 的 mRNA 表达。(J) 平滑的曲线拟合显示 DNMT1 表达与 OSCC 患者死亡风险之间的相关性。(K) DNMT1 表达与 OSCC 患者总体生存风险比之间存在相关性。
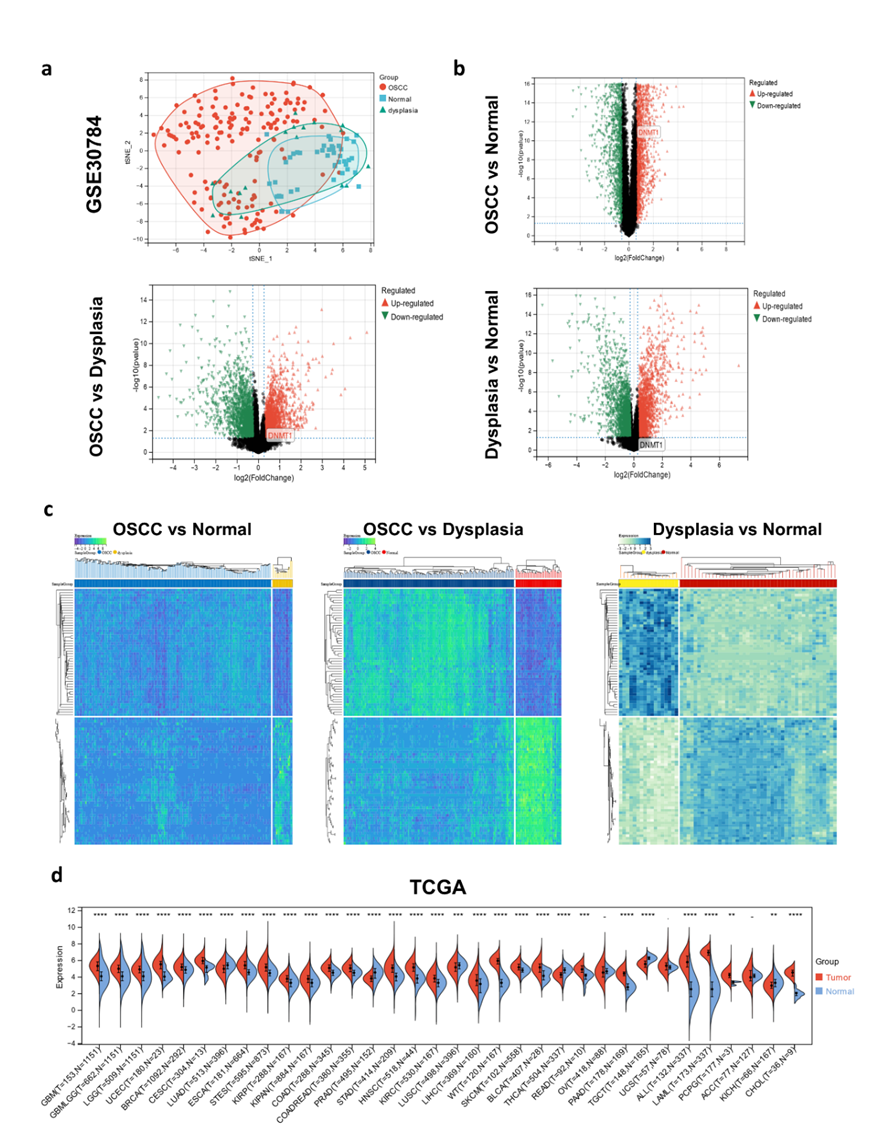
图S2. 基于GEO数据集和TCGA数据库的DNMT1 mRNA表达分析。
(a)GSE30784数据集归一化后的样本主成分分析(PCA)。(b)火山图显示所有差异表达的mRNA。(c)热图显示了前50个显著上调和下调的鳞状细胞癌与正常、鳞状细胞癌与非典型增生、非典型增生与正常的温度。(d)基于TCGA数据库的DNMT1mRNA泛癌表达分析。
02
DNMT1 抑制剂抑制 OSCC 细胞增殖
为了进一步验证 DNMT1 靶向 OSCC 的潜力,他们选择了成熟的 DNMT1 抑制剂,即 GSK-3484862 和 GSK-3685032,进行平行实验,以确定 DNMT1 抑制在 OSCC 细胞生物学行为(主要是增殖和凋亡)中的作用。经过一周不间断的干预后,两种抑制剂均持续降低 OSCC 细胞中的 DNMT1 表达(图 2 A)。在不同浓度下,两种抑制剂均降低了 OSCC 细胞中增殖标志物 Ki67 的表达并增加了凋亡标志物裂解胱天蛋白酶 3 (CC3) 的表达,这与 DNMT1 沉默的影响一致(图 2 A-C)。从功能上讲,两种抑制剂均有效抑制了 OSCC 细胞的自我更新能力(图 2 D-E)。这些结果进一步证实了 DNMT1 是抑制 OSCC 细胞恶性行为的有希望的靶点。
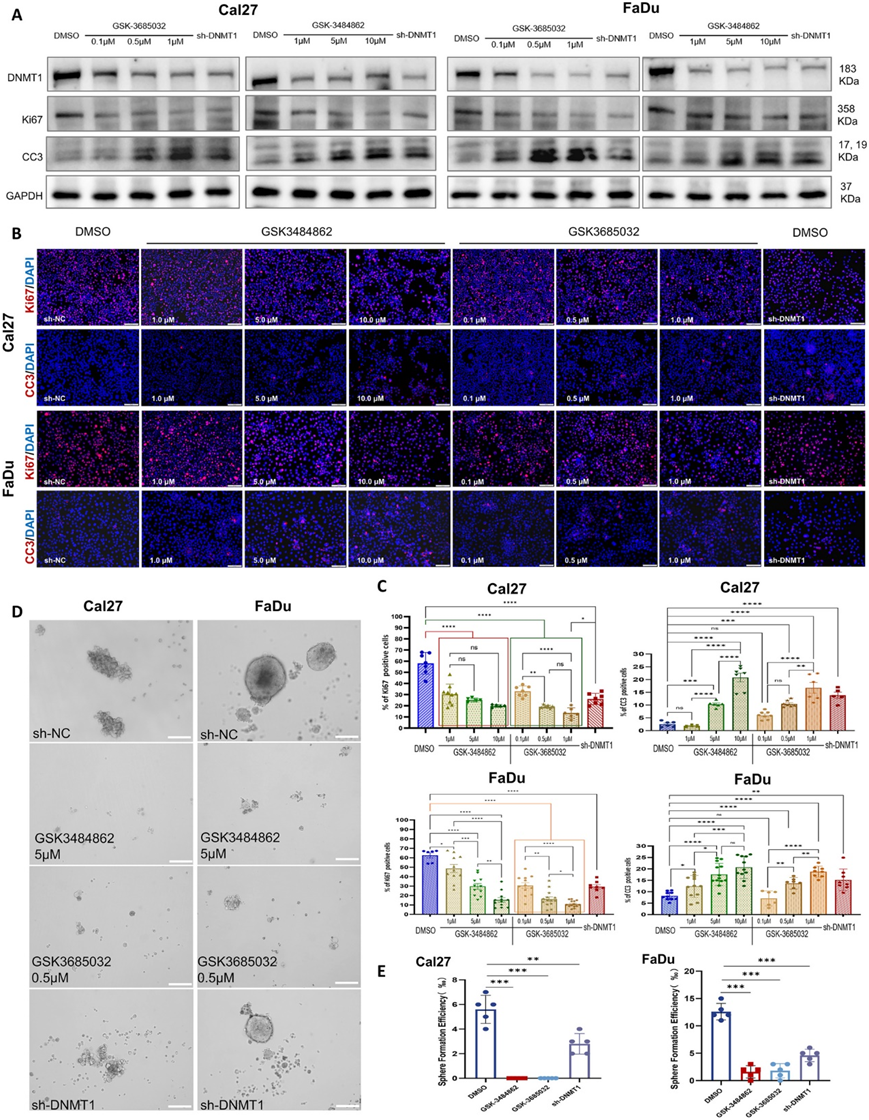
图2. DNMT1 抑制剂持续显著抑制 OSCC 细胞增殖并促进其凋亡。
(a) 免疫印迹分析。 (b-C) 免疫荧光和统计定量。(d-e) OSCC 细胞的球体形成试验和统计定量。
03
全基因组 DNA 低甲基化发生在口腔致癌过程中,并在 OSCC 中稳定维持为癌症特异性稳态
DNMT1 变异直接诱导全基因组 DNA 甲基化的变化。因此,他们通过检测 5-甲基胞嘧啶 (5-mC) 检测了一组新鲜采集的人类样本中的 DNA 甲基化,这些样本包括口腔正常组织、增生性和发育不良性病变以及 OSCC 组织,5-mC 是胞嘧啶第五个碳原子上的共价甲基化是整体 DNA 甲基化的特异性标记。值得注意的是,发育不良和 OSCC 组织的 5-mC 表达均显著低于正常组织,而正常和增生组织之间没有显著差异(图 3 A)。
为了深入研究全基因组 DNA 低甲基化,他们采用 850 k 芯片来确定 OSCC 细胞的整体 DNA 甲基化状态(图 S3A-B)。与正常细胞相比,OSCC 细胞表现出显著差异的 DNA 低甲基化位点,高甲基化位点数量约为 2.16 倍(图 3 B)。这两种细胞类型之间的差异 CpG 位点分布与 850 k 微阵列上的 Infinium 探针分布相似(图 3C-D,图S3C-D)。大多数差异甲基化位点(DMS)位于 CpG 岛内,而不是相邻的岛周围,即岛屿 2 kb 以内的区域(图 3 C,图S3 D)。当使用基因区域探针时,大多数 DMS 位于基因体区域内,而不是启动子内(图 3 D;图S3 D)。对前 3000 个显著 DMS 的 β 值的分析显示,与 NOK 细胞相比,低甲基化程度更高(图 3 E)。对前 1000 个显著 DMS 的分析表明,一小部分显著的高甲基化位点发生在 OSCC 细胞中,其中大多数位于 CpG 岛内(图 3 F)。总之,这些 DNA 甲基化微阵列数据表明 OSCC 细胞中存在癌症特异性低甲基化状态。这种内部低甲基化稳态可能与癌细胞存活的维持有关,这通过 GO 分析中细胞间信号传导、细胞群体增殖和运输和细胞分化调节等生物过程的富集(图 3 G,图S3 E)和信号转导通路的参与得到证明,例如根据 KEGG 分类的PI3K-AKT、MAKP 和 Rap1 信号通路(图 S3 E)。
为了更好地了解这些 DMG 的潜在功能,对 Cal27 和 NOK 细胞进行了额外的 RNA-seq(图S3 F),发现 979 个上调基因和 629 个下调基因,(图 S3 G)。当重叠来自 850k 芯片的这些 DMG、来自细胞 RNA 测序和 TCGA 数据库的 DEG 时,标志基因集富集分析确定了与上皮恶性肿瘤和癌症进展密切相关的途径,包括 Hedgehog、Hypoxia、PI3K-AKT-mTOR、P53 和上皮-间质转化(图 3 H)。
此外,他们使用 GSE204943 数据集分析了整体 DNA 甲基化,该数据集包含正常组织、OLK 病变和 OSCC 组织的数据。虽然 OLK 病变中的 DNA 甲基化水平与正常样本相比没有下降,但在 OSCC 组织中观察到显著降低(图3 I)。这可能是因为该数据集中的 OLK 病变在病理上未细分为增生或发育不良。对 TCGA 数据库的进一步分析证实 OSCC 组织中的整体 DNA 甲基化低于邻近的正常组织(图3 J)。此外,平滑曲线拟合显示整体 DNA 甲基化与死亡风险之间存在非线性和动态关联。 DNA 甲基化水平越高,OSCC 患者的死亡风险就越低(图3 K)。风险比与整体 DNA 甲基化之间存在非线性关联,风险比降低时,整体 DNA 甲基化会显著改变(无论是显著降低还是增加),从而提高生存率(图3 L)。
综合起来,这些结果强烈表明,口腔恶性转化过程中观察到的渐进性全基因组低甲基化与健康细胞中观察到的典型甲基化模式不同。然而,癌细胞似乎能够维持特定于其恶性表型的独特甲基化稳态,这可能导致其不受控制的生长。针对这种癌症特异性甲基化稳态可能代表改变癌细胞增殖的潜在干预措施。
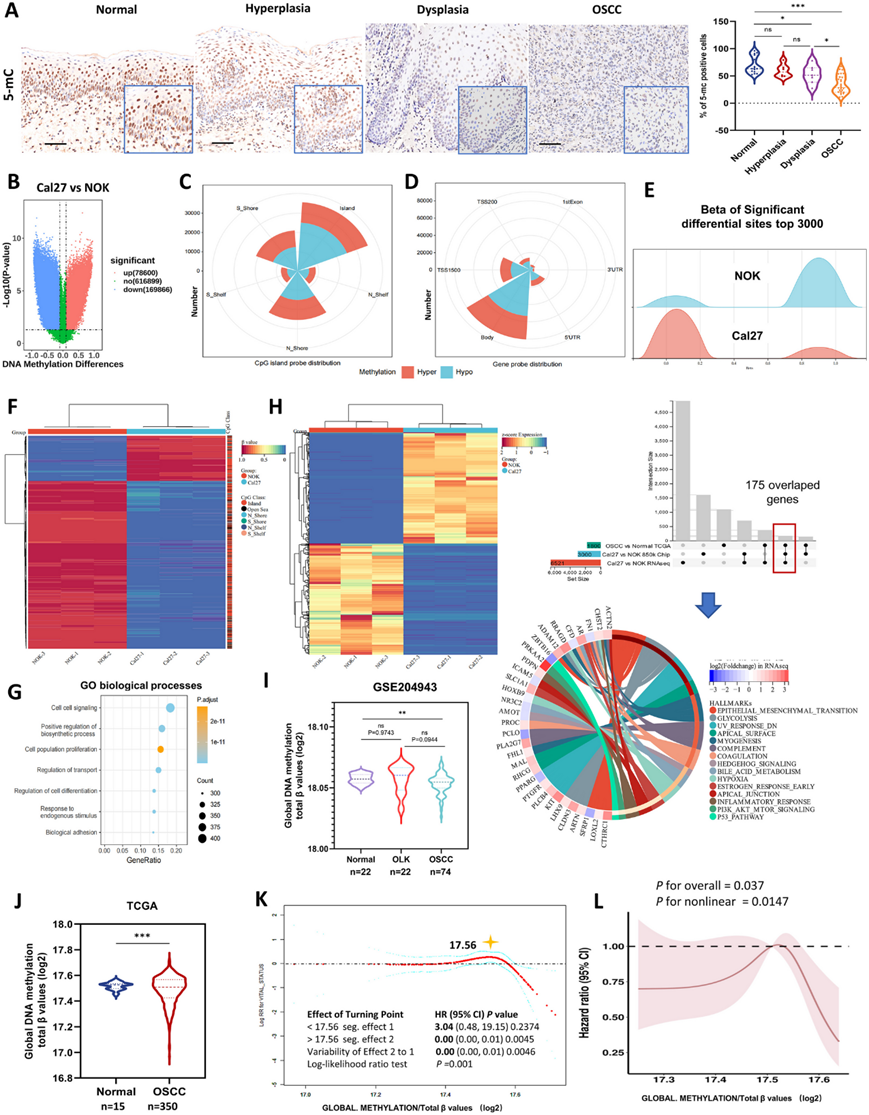
图S3. OSCC癌细胞和NOK细胞的850k芯片分析。
(a)样本PCA分析。(b) Ridgeline图显示了所有样本的总归一化β值分布。(c) 南丁格尔玫瑰图显示了所有850K甲基化位点的探针分布。(d) 直方图分别显示CpG岛探针分布和基因探针分布的所有显著dms的比例。(e) Cal27和NOK细胞GO过程及显著差异甲基化基因Top 3000的KEGG富集分析 (f-g) UMAP和火山图显示Cal27和NOK细胞在RNA-seq中的显著差异。
04
DNMT1 敲低重塑了 OSCC 中高度破坏的全基因组 DNA 低甲基化模式
如上所述,OSCC 组织和细胞系表现出 DNMT1 过表达以及整体 DNA 低甲基化。此外,沉默 DNMT1 可有效抑制肿瘤生长。随后在 DNMT1 敲低小鼠产生的萎缩 OSCC 肿瘤中检测 5-mC 发现 5-mC 表达显著下降(图4 A)。这一发现表明,取决于 DNMT1 的低甲基化状态与肿瘤抑制有关。
为了进一步了解 DNMT1 如何重塑 OSCC 中的整体 DNA 低甲基化,他们使用 DNA 甲基化 850 k 芯片将 sh-DNMT1 癌细胞与 sh-NC 细胞进行了比较。令人惊讶的是,sh-DNMT1 癌细胞表现出广泛且几乎完全的全基因组 DNA 低甲基化。这从 358933 个差异低甲基化位点的鉴定中可以看出,与 sh-NC 癌细胞相比,差异高甲基化位点的数量约为 61.02 倍(图4B)。前 3000 个显著 DMS 的 β 值分布导致 OSCC 细胞中的 DNA 甲基化几乎完全消除(图4C)。这些发现进一步证明 DNMT1 是调节 DNA 甲基化的必需酶;DNMT1 活性的任何改变或中断都与 DNA 甲基化的变化密切相关。
他们接下来分析 DNMT1 重塑的整体 DNA 低甲基化模式图片。DNMT1 靶向引起的大多数差异低甲基化位点位于 CpG 岛周围而非 CpG 岛内(图4 D,图S5A)。OSCC 细胞和 NOK 细胞之间主要探针(图 S3 C)和 DMS 分布的观察差异显著(图S5 B)。当评估前 1000 个 DMS 时,sh-DNMT1 癌细胞表现出完全的 DNA 低甲基化,少数 DMS 分布在 CpG 岛但位于岛周围(图4 E)。功能基因区域分布分析显示,DMS 主要位于基因体内(图4 F,图 S5A),而前 3000 个 DMS 主要位于基因体内,其次是转录起始位点 1500 (TSS1500)区域(图S5 B)。与 NOK 细胞相比,sh-DNMT1 癌细胞也表现出比 sh-NC 细胞更明显的低甲基化;这些差异性低甲基化位点大部分位于 CpG 岛周围和基因体内(图S5 B)。
根据这些结果,可以得出结论,DNMT1 的下调广泛破坏了整体 DNA 甲基化的稳态,并导致 OSCC 细胞直接和持续减少;这种低甲基化的特点是高度分散的分布转变模式。由于这些异常的改变,癌细胞可能难以维持增殖。两组 DMG 都与各种生物过程有关,包括细胞群体增殖、凋亡过程和特定信号转导,其中 PI3K-AKT 最为突出(图4 G,图S5C)。然后他们检测了 PI3K-AKT 通路中的甲基化改变,发现虽然与 NOK 细胞相比,癌细胞中相关的 DMS 略微低甲基化(图4 H),但 DNMT1 敲低导致关键基因的广泛低甲基化(图4 I)。与 PI3K-AKT 信号通路激活有关的关键基因,如PIK3CD、PIK3R1和AKT1,表现出动态低甲基化;相反,抑制基因PTEN表现出与 NOK 细胞中观察到的模式一致的模式(图4 J)。总之,上述发现表明,DNMT1 敲低可以直接重塑 OSCC 细胞中的整体 DNA 低甲基化,导致 PI3K-AKT 信号传导改变后出现普遍的疾病但纯粹的全基因组 DNA 低甲基化模式。
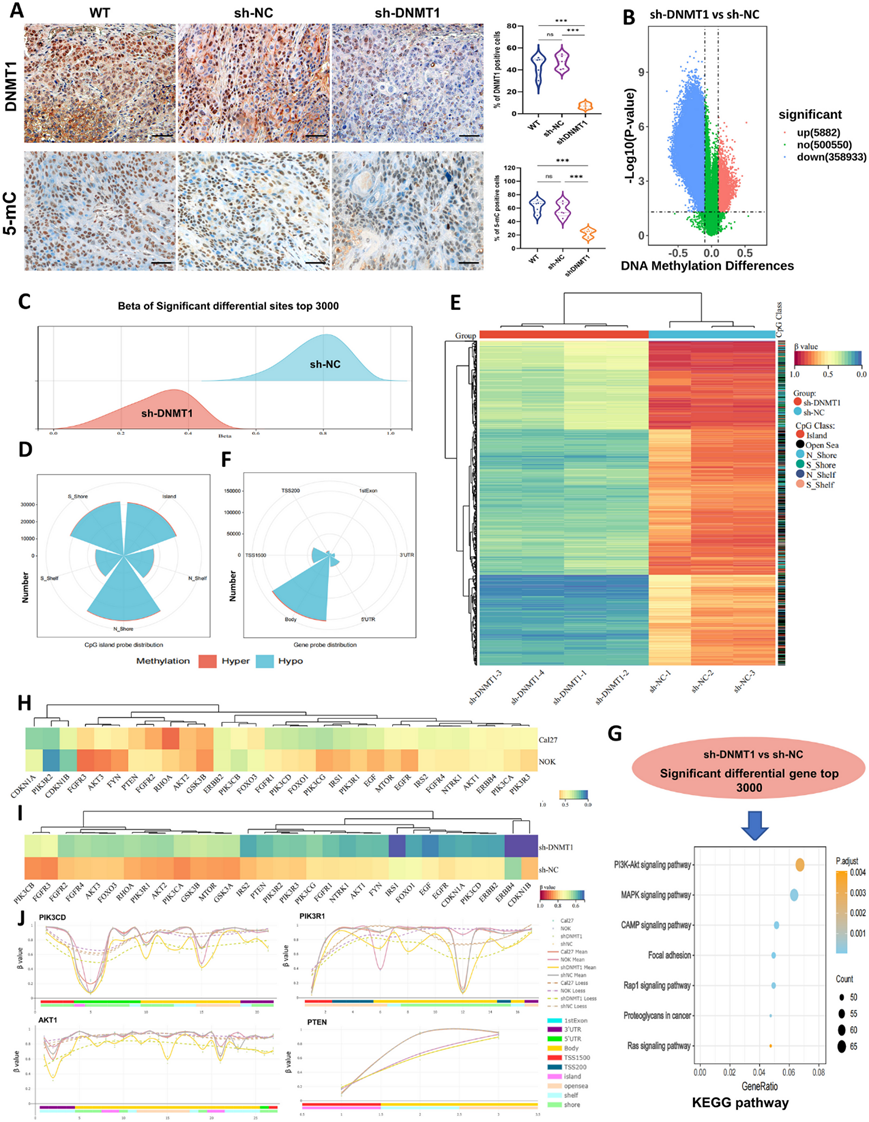
图4. DNMT1 靶向重塑了 OSCC 中广泛而纯粹的全基因组 DNA 低甲基化模式。
(a) 异种移植 OSCC 肿瘤中 DNMT1 和 5-mC 的代表性 IHC 图像和分析。 (b) DNA甲基化 850 k 芯片的火山图显示 sh-DNMT1 细胞与 sh-NC Cal27 细胞相比存在显著差异的 DNA 甲基化位点。(c) 岭图显示 sh-DNMT1 和 sh-NC Cal27 细胞中前 3000 个显著差异位点的 β 值分布。(d) 南丁格尔玫瑰图显示具有 CpG 岛探针的所有 DMS 的分布。(e) 热图显示具有 β 值绝对差异的前 1000 个差异 CpG 位点。(f) 南丁格尔玫瑰图显示具有基因探针分布的所有显著 DMS 的比例。(g) 气泡图展示DNA甲基化位点相关DEGs的KEGG富集情况。(h-i) 热图展示Cal27与NOK细胞、sh-DNMT1与sh-NC癌细胞之间PI3K-AKT通路基因差异甲基化位点的平均甲基化水平。(J) 所有显著的DMS都在基因PIK3CD、PIK3R1、AKT1和PTEN中富集,它们是PI3K-AKT通路中的代表性关键基因。
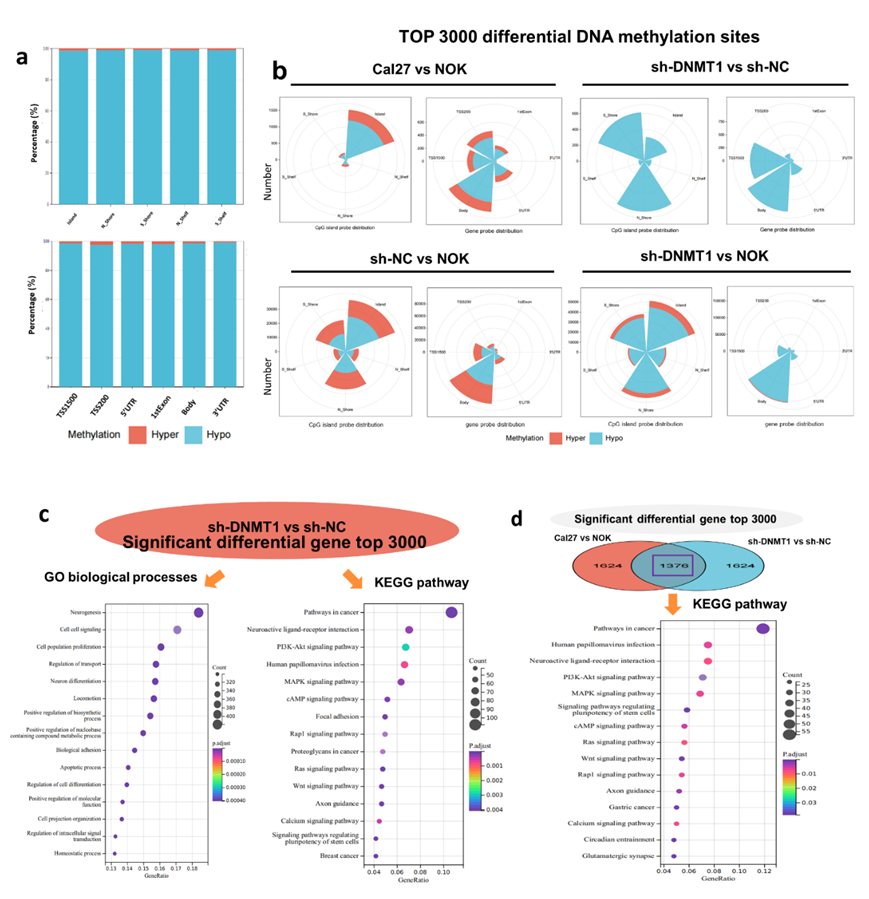
图S5. DNMT1靶向重塑了癌细胞的整体DNA甲基化,并与PI3K-AKT通路相关。
(A) 直方图分别显示CpG岛探针分布和基因探针分布的所有显著dms的比例。(B) 南丁格尔玫瑰图显示根据CpG岛探针分布和基因探针分布在不同细胞类型中的显著dms数Top 3000。 (C)GO分析。(D) 从Cal27与NOK细胞和sh-DNMT1与sh-NC癌细胞中筛选出的Top 3000重叠DME的KEGG富集分析。
05
DNMT1 重塑的整体 DNA 低甲基化介导 PI3K-AKT 抑制并增强肿瘤生长抑制
通过KEGG分类分析,他们确定了PI3K-AKT信号通路是与DNMT1特异性DNA甲基化模式相关的重要通路。Cal27细胞与NOK细胞、sh-DNMT1细胞与sh-NC Cal27细胞之间前3000个DMG的重叠进一步支持了这一发现(图5 A,图S5D)。鉴于与PI3K-AKT信号通路相关的DMS,所有这些DMS在DNMT1敲低后都发生了低甲基化,并且大多数位于CpG岛周围和基因体内,尤其是AKT相关的DMS(图S6B)。这一发现表明PI3K-AKT的激活可能受到抑制,因为基因体内的甲基化水平和发生在CpG岛周围的肿瘤特异性甲基化通常与基因表达呈正相关。
然后,他们评估了总蛋白和磷酸化蛋白的表达水平,结果显示 sh-DNMT1 癌细胞中磷酸化 AKT 和 mTOR 显著降低,但是PI3K 和 AKT 的整体蛋白表达水平没有表现出实质性的改变(图5B)。这意味着由于 DNMT1 重塑的 DNA 低甲基化,PI3K-AKT 的激活受到显著阻碍。在 sh-DNMT1 细胞中使用 PI3K 激动剂 (740 YP) 进行了挽救实验。这些结果表明 740 YP 能够部分恢复 sh-DNMT1 细胞中 mTOR 的磷酸化水平(图5B)。
已证实 PI3K 抑制剂可抑制多种 SCC 中的肿瘤生长。然后,他们利用异种移植 OSCC 小鼠模型进行了体内研究,其中 PI3K 由BEZ235 抑制(图5C)。在抑制 DNMT1 和 PI3K 后均观察到肿瘤生长减少(图5D)。有趣的是,与 PI3K 抑制剂治疗相比,DNMT1 敲低对肿瘤缩小的抑制作用更为明显。当将 PI3K 激动剂应用于 DNMT1 沉默的小鼠时,仅观察到肿瘤生长略有增加(图5D)。在 sh-DNMT1 组中观察到增殖细胞数量减少和凋亡细胞数量增加(图5E)。此外,DNMT1 敲低有效阻断了皮下肿瘤中的 PI3K-AKT 信号通路,AKT 和 mTOR(p-AKT 和 p-mTOR)磷酸化降低表明了这一点。用 PI3K 激动剂治疗后,PI3K-AKT 信号转导得到部分恢复,但与 sh-NC 组相比受到抑制(图5E)。他们检测了人类样本中 DNMT1 或 5-mC 和 p-AKT 的共定位,包括正常、发育不良和 OSCC 组织。在口腔致癌过程中,观察到 p-AKT 增加,随后是 DNMT1 过度表达和 5-mC 表达丧失(图5F)。
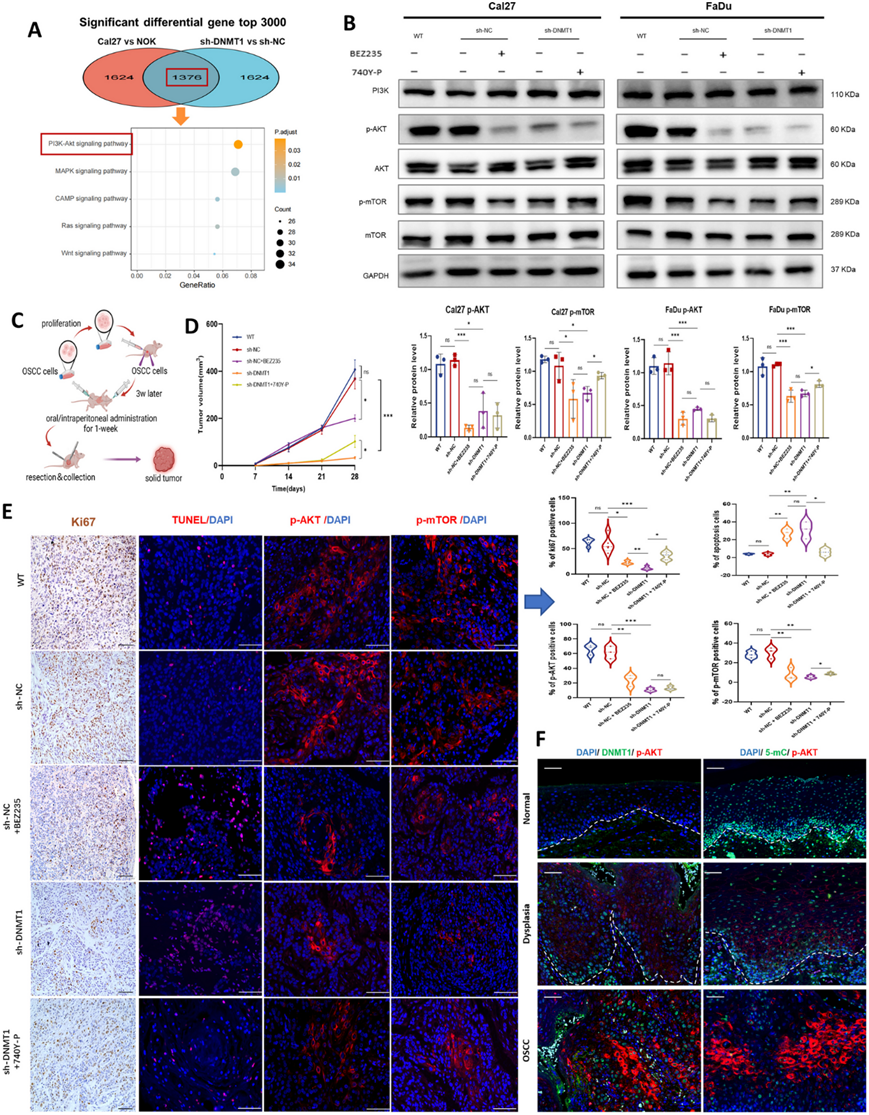
图5. PI3K-AKT 通路参与 DNMT1 重塑的 DNA 低甲基化模式,从而调节口腔肿瘤转化和肿瘤生长。
(a) 气泡图显示 Cal27 和 NOK 之间以及 sh-DNMT1 和 sh-NC 细胞之间差异 DNA 甲基化位点相关的前 3000 个重叠基因的 KEGG 富集情况。(b)免疫印迹分析。(c) 示意图显示异种移植 OSCC 模型。(d) 肿瘤生长曲线。(e) 的代表性染色图像和量化。(f) 包括正常、发育不良和 OSCC 组织在内的口腔人类样本中三个通道(即 DAPI、DNMT1 或 5-mC 和 p-AKT)的代表性 mIHC 图像。
06
DNMT1 靶向作用可通过额外抑制 CDK2-Rb 通路实现更好的肿瘤抑制
为了确定 DNMT1 抑制和由此导致的 DNA 甲基化改变的其他分子机制,他们使用 TCGA 数据库和 4 个 GEO 数据集进行了交叉meta分析,然后与850 k 芯片配置文件的与 DNMT1 相关的 DEG 重叠。该分析确定了总共 152 个与 DNMT1 和整体 DNA 甲基化相关的差异表达基因(DEG)。随后通过 KEGG 富集分析验证了这些 DEG 与 DNA 复制、细胞周期、碱基切除修复和凋亡途径相关。通过蛋白相互作用(PPI)分析和实施网络过滤器,他们确定了一组五个不同的节点分子,即 CDK2、GSK3B、ABL1、NFKB1 和 CREBBP。与正常组织相比,TCGA 数据库中的 OSCC 样本中只有 CDK2 和 GSK3B 的 mRNA 表达显著增加,两者均与 OSCC 中的 DNMT1 表达呈正相关(图 S7E)。
CDK2 通过调节细胞周期和磷酸化视网膜母细胞瘤蛋白 (Rb),在癌症进展中发挥关键作用,尤其是在 G1-S 期过渡期间。与 sh-NC 细胞相比,sh-DNMT1 癌细胞中 CDK1/2/3 和 Rb 的磷酸化水平降低(图6 A),证实了 CDK2-Rb 信号转导受到额外抑制。此外,PI3K 抑制剂已证明可在 CDK2-Rb 的失活中发挥作用,并且 PI3K 和 CDK2 抑制剂(BEZ235 和 AT7519)的联合使用具有最明显的抑制作用,表明 PI3K-AKT 和 CDK2-Rb 信号通路之间存在潜在的相互作用。然而,PI3K 激动剂不能完全恢复 CDK2-Rb 的激活,这意味着 DNMT1 介导的 CDK2-Rb 和 PI3K-AKT 抑制可能同时发生。此外,他们观察到肿瘤转化过程中 p-Rb 水平持续增加,同时 DNMT1 表达升高,5-mC 减弱(图6B),证实了 DNMT1-DNA 甲基化模式对 CDK2-Rb 通路的调节作用。
他们进一步对异种移植小鼠进行了 BEZ235 和 AT7519 的联合治疗,靶向 PI3K-AKT 和 CDK2-Rb 通路的双重抑制,作为阳性对照。用联合抑制剂治疗的小鼠表现出与携带 sh-DNMT1 肿瘤的小鼠相当的肿瘤生长抑制。与接受单一 PI3K 抑制剂的小鼠相比,DNMT1 沉默的小鼠在早期干预中也表现出最慢的肿瘤发生和对肿瘤生长的更好控制(图6C)。在研究结束时,与携带 sh-NC 肿瘤的小鼠相比,接受干预的所有三组携带肿瘤的小鼠的肿瘤体积均显着缩小。然而,sh-DNMT1 组与其他两组用抑制剂治疗的组相比,并没有表现出更好的肿瘤抑制效果(图6D-E)。这种缺乏优势可能归因于 DNMT1 的可逆性质,它可以抵消 DNMT1 基因沉默的影响。然而,他们确实观察到这三个实验组中细胞增殖减少和细胞凋亡增加,为这些干预措施的抗癌效果提供了有力的证据。他们发现 sh-DNMT1 肿瘤小鼠中 Ki67 阳性细胞的比例有所降低,这为 DNMT1 沉默增强的抗癌效果提供了进一步的证据(图6F)。此外,sh-DNMT1 组中 Rb 蛋白的磷酸化降低(图6 F),证明DNMT1 重塑的 DNA 低甲基化可以通过同时抑制 PI3K-AKT 和 CDK2-Rb 信号通路来抑制肿瘤生长。
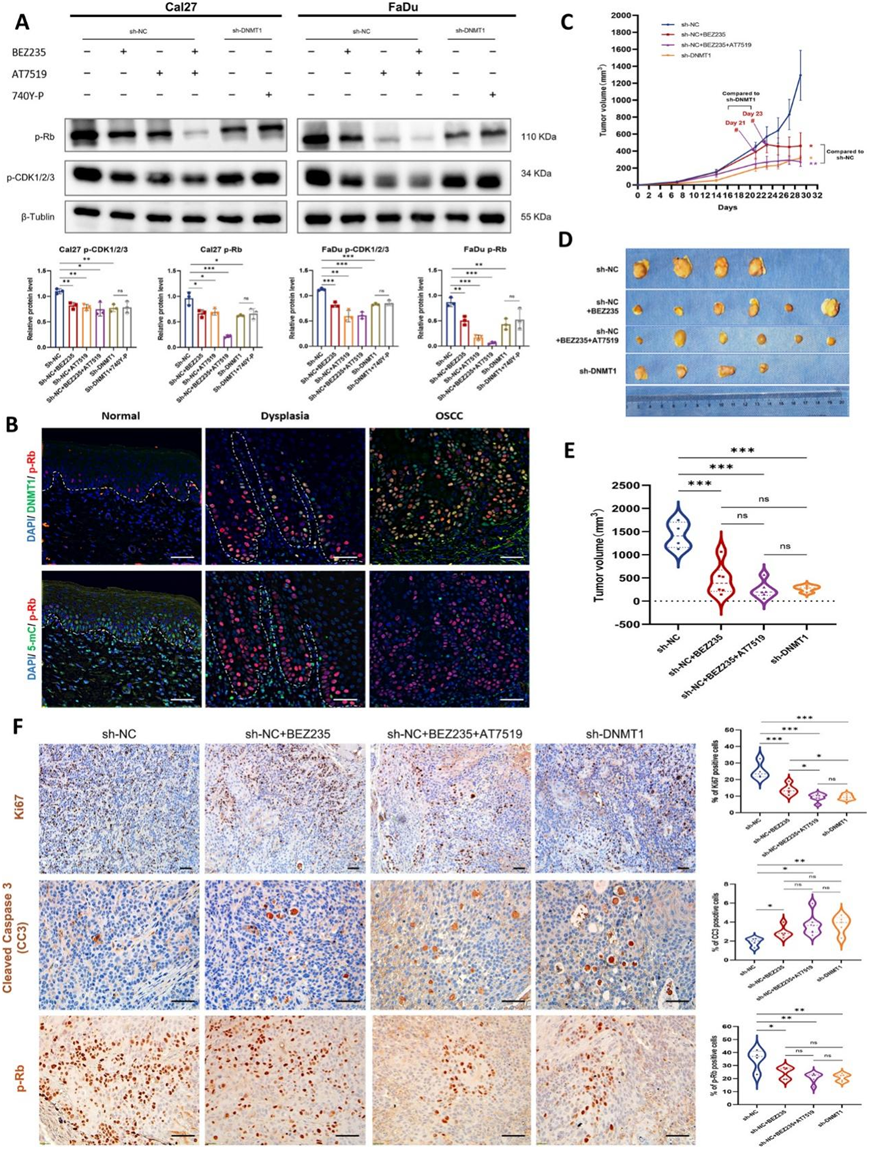
图6. 抑制 CDK2-Rb 信号通路有助于增强由 DNMT1 重塑的整体 DNA 低甲基化引起的肿瘤抑制。
(a)免疫印迹分析。(b) 代表性 mIHC 图像。(c) 肿瘤生长曲线。(d-e) 研究结束时大体异种移植肿瘤和肿瘤体积统计的呈现。(f) 异种移植 OSCC 肿瘤中 Ki67、裂解胱天蛋白酶 3 (CC3) 和 p-Rb 的代表性IHC图像和分析。
07
DNMT1 敲低进一步诱导 GSK3β 失活,促进细胞凋亡
关于与 DNMT1 介导的 DNA 低甲基化密切相关的另一个候选基因 GSK3B,显示异种移植肿瘤中 Ser9 位点的 GSK3β 磷酸化 (p-GSK3β) 发生了显著变化。当 DNMT1 敲除或同时应用 PI3K 和 CDK2 抑制剂时,p-GSK3β 显著增加。相反,PI3K 抑制导致 p-GSK3β 明显减少(图7 A)。GSK3β 是维持细胞代谢平衡的重要调节酶,尤其是在癌细胞中的糖原合成和糖酵解等过程中。其失活是通过 Ser9 的磷酸化实现的,从而促进葡萄糖合成。随后,他们检测了 sh-DNMT1 肿瘤组织中糖原聚集的增加,与用联合抑制剂治疗的肿瘤组织相似,但明显高于仅用 PI3K 抑制剂或载体治疗的肿瘤。此外,PI3K 抑制剂治疗的肿瘤表现出很少的糖原积累(图7 A)。DNMT1 敲低和 PI3K 和 CDK 联合抑制都导致肿瘤组织中糖酵解水平升高,如 PFK 和 PKM2 阳性细胞数量增加所示。这些细胞高度位于糖原沉积物较多的区域(图7A-B)。有趣的是,这些表现出糖原聚集和糖酵解增强的细胞主要位于凋亡肿瘤区域附近,远离增殖区域(图7 B)。综上所述,这些结果表明 DNMT1 诱导的 GSK3β 失活通过异常的糖原代谢促进癌细胞死亡。
虽然 GSK3β 通常受 AKT 信号调控,但体外实验表明,与单一 PI3K 抑制相比,靶向 DNMT1 和联合抑制 PI3K 和 CDK2 均可逆转 OSCC 细胞中 p-GSK3β 表达的增加。有趣的是,只有 DNMT1 敲低才会持续抑制 PI3K-AKT 活化(图7 C)。相反,单一 CDK2 抑制剂对 AKT 磷酸化有刺激作用,而联合 CDK2 抑制剂对 PI3K-AKT 活化没有影响。这些发现还表明,DNMT1 沉默可以通过同时失活 PI3K-AKT、CDK2-Rb 和 GSK3β 来恢复信号平衡。此外,在人类口腔多发癌样本中,观察到 p-GSK3β 表达的广泛变化以及 p-AKT 和 DNMT1 表达和 DNA 甲基化的变化(图7D)。
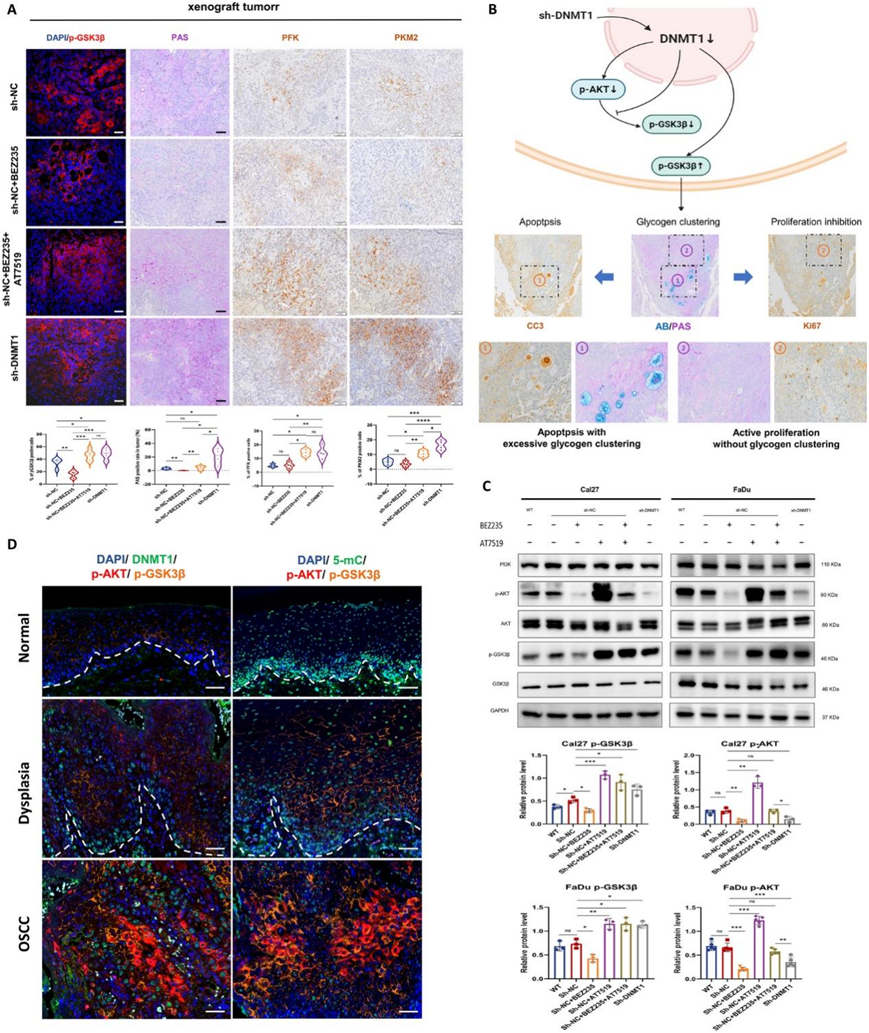
图7. DNMT1 敲低导致 GSK3β 失活,从而导致肿瘤中糖原过度聚集和细胞凋亡。
(a) 异种移植 OSCC 肿瘤中 p-GSK3β 的代表性 IF 图像和糖原、PFK 和 PKM2 的 PAS/IHC 图像。(b) DNMT1 敲低通过上调 p-GSK3β 阻止 PI3K 抑制下游的 GSK3β 激活,导致肿瘤中糖原过度聚集(AB-PAS 染色),位于细胞凋亡周围,远离增殖(IHC 染色)。(c) 免疫印迹分析。(d) 代表性 mIHC 图像显示口腔人类样本(包括正常、发育不良和 OSCC 组织)中的 DAPI、DNMT1 或 5-mC、p-AKT 和 p-GSK3β。
从动物癌症模型和临床试验中可以看出,PI3K 抑制剂会导致全身葡萄糖紊乱。这种紊乱会限制抗癌疗法的有效性。在 OSCC 小鼠模型中,他们进行了实验以观察血糖水平的变化(图8 A)。施用 PI3K 抑制剂会逐渐引起高血糖症。相反,当 PI3K 抑制剂与 CDK2 抑制剂联合使用时,血糖水平会迅速显著下降 (图8B-D)。在治疗后 5 小时的时间点,用 BEZ235 治疗的小鼠的血清胰岛素水平保持持续升高,而用联合抑制剂治疗的小鼠的血清胰岛素水平急剧下降(图8 E)。此外,以 DNMT1 为靶点的肿瘤小鼠的血糖和血清胰岛素水平都表现出显著的稳定性。
先前的研究表明,PI3K 抑制引起的暂时性高血糖通常会持续数小时,因为胰岛素反馈机制会启动以恢复正常的血糖稳态。因此,他们检测了肝脏中的糖原合成情况,发现只有用 PI3K 抑制剂治疗的荷瘤小鼠表现出明显高的糖原水平(图8F-G),同时伴有 p-GSK3β 表达升高(图8F-H)。相反,因联合抑制剂治疗而出现低血糖的小鼠表现出肝糖原储存减少和 p-GSK3β 水平降低,进一步表明肝糖原分解有助于恢复正常血糖。然而,sh-DNMT1 肿瘤小鼠的 p-GSK3β 水平略高于未经治疗的异种移植小鼠,这表明 sh-DNMT1 肿瘤本身的糖原消耗也可能在一定程度上引发肝糖原代偿性代谢。总之,这些发现表明,针对 DNMT1 的抑制干预最有可能降低不良毒性,从而在 PI3K 抑制引起的高血糖情况下维持正常的血糖平衡,进一步增强抗癌治疗的有效性。
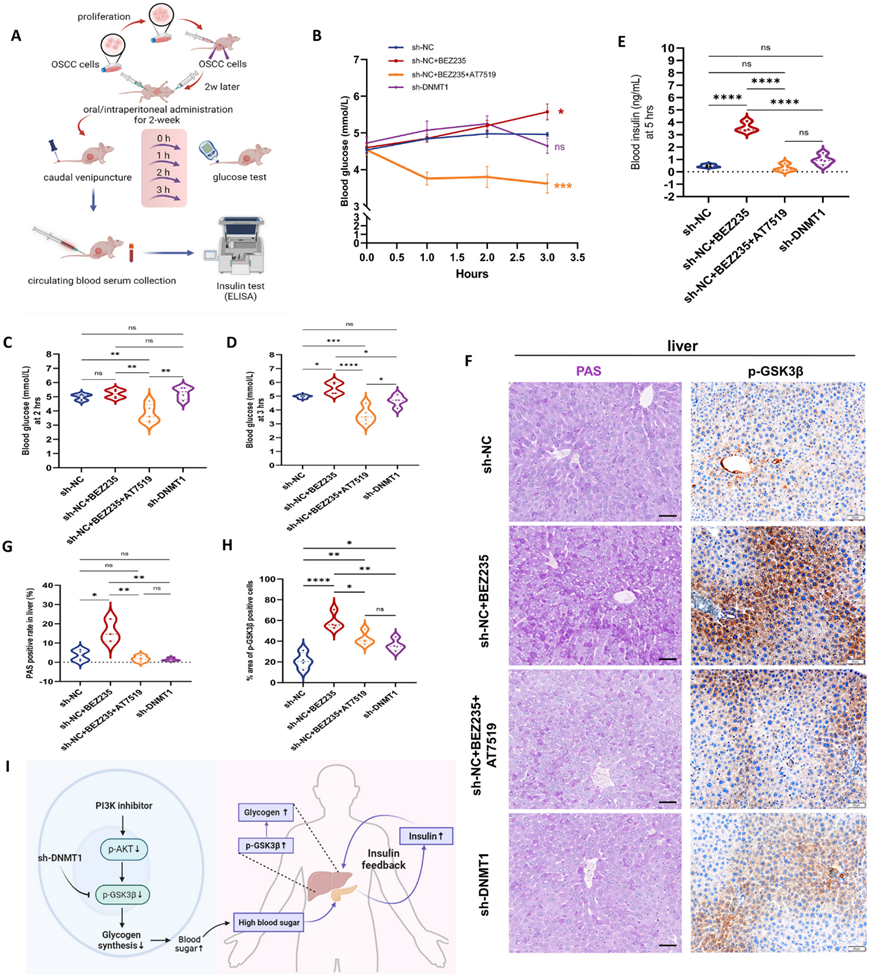
图8. DNMT1 敲低导致的 GSK3β 失活可拮抗 PI3K 抑制引起的胰岛素反馈。
(a) 肿瘤小鼠血糖和血清胰岛素检测实验示意图。(b) 给药后 3 小时内血糖折线图。(c-d)血糖变化情况。(e) 给药后 5 小时小鼠的血清胰岛素水平。(f-h) 给药后 5 小时小鼠肝脏中糖原/p-GSK3β 的代表性 PAS/IHC 图像和分析。(i) PI3K 抑制剂通过去磷酸化激活 GSK3β,阻碍糖原合成并导致有害的高血糖效应。
08
DNMT1介导的口腔致癌及抗癌作用的信号协同模式及机制图
口腔恶性转化和OSCC治疗过程中可能存在PI3K-AKT、CDK2-Rb和GSK3β参与的协同信号转导通路,且这些通路受DNMT1-DNA甲基化模式的影响。OSCC是一种异质性实体肿瘤,从正常病变到癌前病变再到癌变病变,经历了多个致癌步骤。为了进一步验证口腔恶性转化过程中的这种协同信号转导模式,他们使用GSE181919数据集进行了单细胞转录组分析,以更好地了解单细胞水平上肿瘤异质性的遗传变异。
他们首先分别从正常、OLK 和 HNSCC 组织中分离出上皮细胞。考虑到上皮细胞群中存在细胞异质性,重新分类二倍体和非整倍体细胞,因为后者可以完全代表恶性群体。伪时间轨迹分析显示正常上皮细胞有两个进展分支,一个导致癌前状态,另一个导致癌变状态,非整倍体细胞显著位于后者(图9 A)。随着上皮细胞向癌变状态发展,协同信号通路(包括 PI3K-AKT、mTOR、CDK-Rb-E2F 和糖酵解)发生激活(图9 B)。对比癌前阶段的趋势,恶性过程中涉及的所有信号转导通路均表现出持续激活(图9B)。此外,对调控该信号协同模式的单个关键基因进行伪时间轨迹分析,证实了DNMT1、AKT1、CDK2和GSK3B在上皮癌变过程中表达增加,而负调控基因PTEN表达降低(图9D)。因此,单细胞转录组分析结果进一步证实了信号协同功能模式在口腔癌变中的关键作用。
在异种移植肿瘤中,他们还利用多重免疫组织化学技术来确定蛋白质功能水平上核心调控标记物的共定位(图9D)。在活跃增殖的 OSCC 肿瘤中,癌细胞表现出高水平的 p-AKT、p-Rb 和 p-GSK3β。大多数癌细胞表现出这三种标记物的频繁共定位,无论它们是高表达还是弱表达(图9D)。在 DNMT1 靶向后,异种移植肿瘤露出分散的较小癌性病变,p-AKT 和 p-Rb 表达降低,但 p-GSK3β 仍然大量表达。特别是,p-Rb 和 p-GSK3β 的改变与 AKT 磷酸化的变化无关(图9D)。这些发现再次证实了 DNMT1 在协调多种信号转导中的强大调节作用。
综合上述发现,他们绘制了一个示意图,阐明了 DNMT1 在口腔恶性转化和控制肿瘤生长中的作用(图9E)。DNMT1 表达逐渐升高,与全基因组 DNA 低甲基化同步,从而引发 OSCC 的多阶段致癌作用。靶向 DNMT1 会导致异常和广泛的 DNA 低甲基化状态的形成,从而诱导特定的信号协同作用。这些途径涉及 PI3K-AKT 和 CDK2-Rb 的双重抑制,以及 GSK3β 的失活,从而调节细胞增殖减少和细胞凋亡增加,从而阻碍肿瘤生长。此外,靶向 DNMT1 可能会抵消 PI3K 抑制引起的高血糖和胰岛素反馈的药理毒性。该过程通过诱导超额 GSK3β 失活来激活,从而导致持续阻断 PI3K-AKT 激活。所有这些过程代表了由 DNMT1 引起信号协同作用,通过提高效率和降低毒性来有效抑制肿瘤生长。
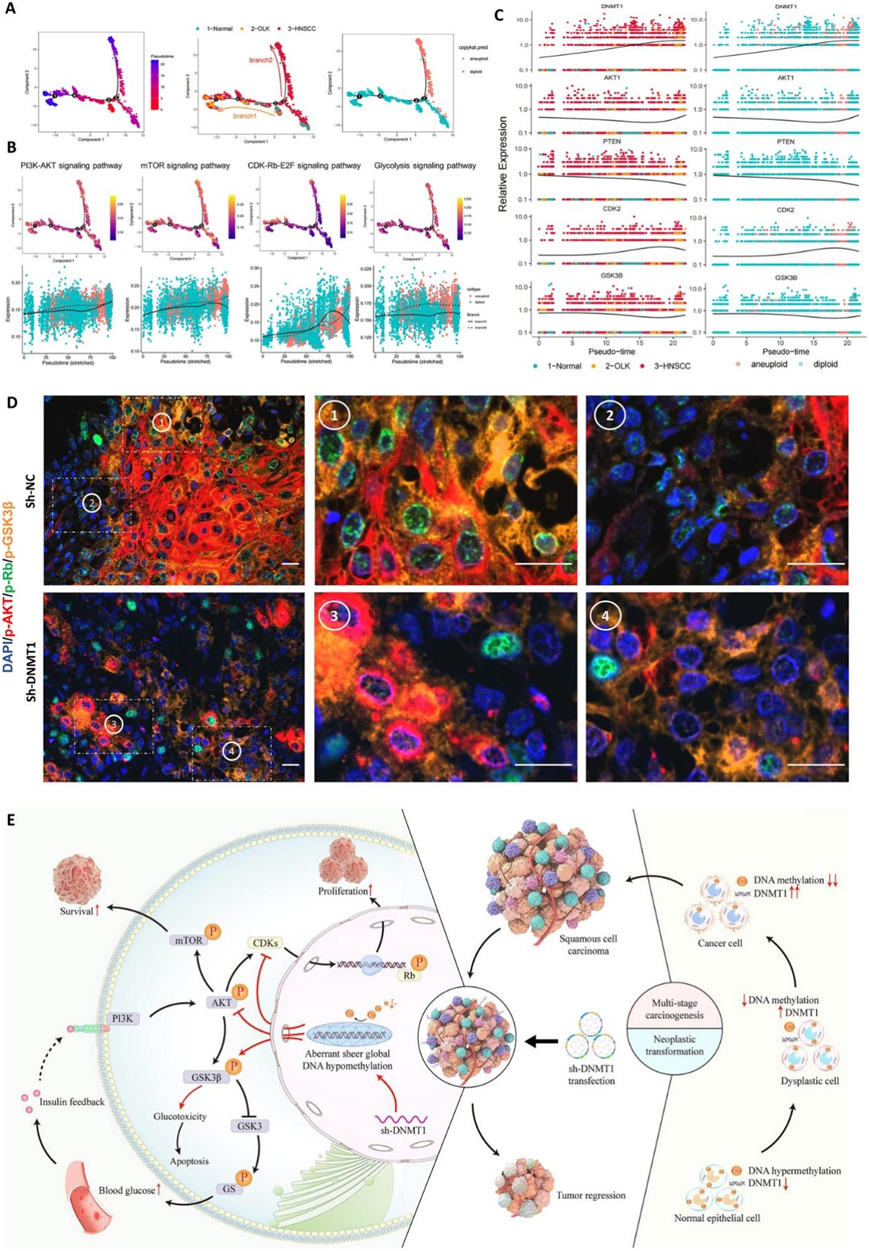
图9. DNMT1 介导的信号协同模式和示意图在口腔致癌作用和抗癌功效中的作用。
(a) 上皮细胞的伪时间轨迹。(b) 上皮细胞的伪时间轨迹。(c) DNMT1、PTEN、AKT1、CDK2 和 GSK3B 表达的伪时间轨迹分析。 (d) 异种移植 OSCC 肿瘤中 DAPI、p-AKT、p-Rb 和 p-GSK3β 的代表性 mIHC 染色图像。 (e) 示意图表明 DNMT1 依赖的整体 DNA 甲基化模式在口腔致癌作用和 OSCC 治疗中发挥作用,以及所涉及的协同信号转导。
+ + + + + + + + + + +
结 论
本项研究探究了在人类样本中口腔恶性转化过程中 DNMT1 表达的稳定增强,抑制该表达可显著降低体外和异种移植 OSCC 模型中的致瘤性。在口腔致癌过程中,DNMT1 过表达伴随着癌症特异性 DNA 低甲基化的积累;相反,DNMT1 敲低会导致癌细胞和异种移植肿瘤中异常广泛的全基因组 DNA 低甲基化。这种新的 DNMT1 重塑的 DNA 低甲基化模式阻碍了 PI3K-AKT 和 CDK2-Rb 的双重激活,并协同灭活了 GSK3β。在治疗 OSCC 小鼠时,靶向 DNMT1 比 PI3K 抑制剂具有更大的抗癌效果,并降低了 PI3K 抑制剂或 PI3K 和 CDK 抑制剂组合引起的血糖变化的毒性以及不利的胰岛素反馈。靶向 DNMT1 可重塑一种新型整体 DNA 低甲基化模式,从而通过平衡信号协同作用提高抗癌效果并最大程度降低潜在毒性作用。
+ + + + +



