

 English
English文献解读|Nat Immunol(27.7):代谢调节因子 PKM2 的缺乏会激活戊糖磷酸途径并产生 TCF1 +祖细胞 CD8 + T 细胞以改善免疫疗法
✦ +
+
论文ID
原名:Deficiency of metabolic regulator PKM2 activates the pentose phosphate pathway and generates TCF1+ progenitor CD8+ T cells to improve immunotherapy
译名:代谢调节因子 PKM2 的缺乏会激活戊糖磷酸途径并产生 TCF1 +祖细胞 CD8 + T 细胞以改善免疫疗法
期刊:Nature Immunology
影响因子:27.7
发表时间:2024.09.26
DOI号:10.1038/s41590-024-01963-1
背 景
CD8+ T 细胞是抗肿瘤免疫反应的关键决定因素。它们的治疗效用已通过患者细胞群和药理学试剂(包括免疫检查点阻断)的过继细胞转移 (ACT) 方法得到充分利用。ACT方法,包括嵌合抗原受体T (CAR -T)细胞疗法,需要体内转移患者自身的淋巴细胞,这些淋巴细胞可以进行扩增、选择和基因改造,以增强其抗肿瘤特异性和功效。TCF1high祖细胞 CD8 + T 细胞介导免疫疗法的疗效;然而,控制其产生和维持的机制尚不清楚。
实验设计
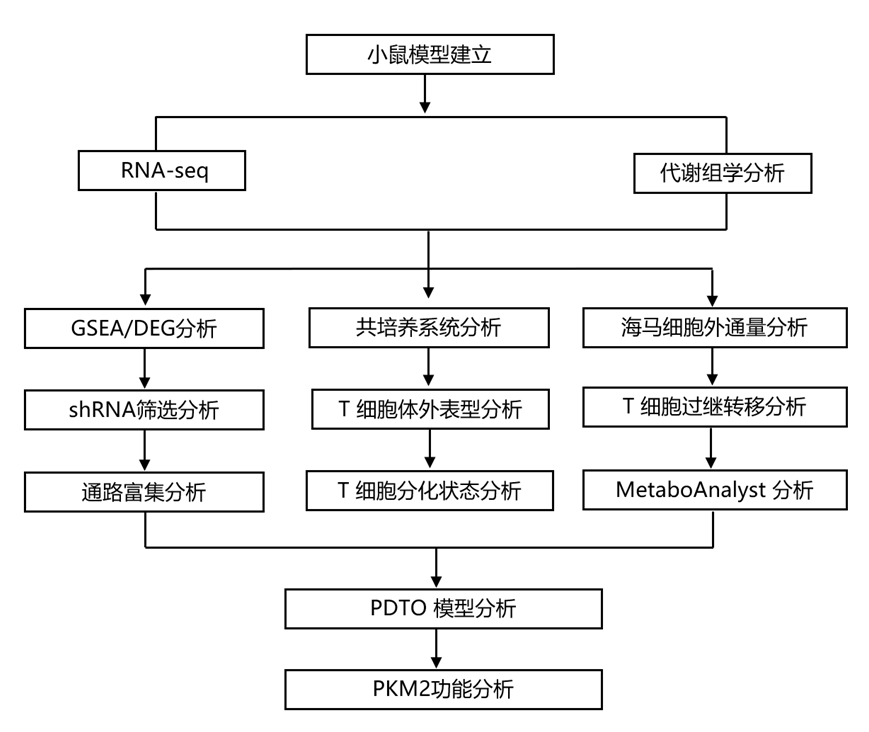
结 果
01

丙酮酸激酶 M 是 CD8+ T 细胞命运的调节因子
为了研究转录组进化与肿瘤进展的关系,研究团队对非小细胞肺癌 (NSCLC) HKP1 原位小鼠模型中的肿瘤浸润性 CD8 + T 细胞(图1a)进行了转录组分析(RNA-seq)。主成分分析 (PCA) 显示样本聚类与时间和肿瘤负荷有关(图1b)。他们整合了公开发表的抗 PD-1 治疗数据集,评估大肿瘤(无反应)和小肿瘤(有反应),以确定干预的分子靶点。通过基因集富集分析(GSEA)鉴定了 343 个差异表达基因(DEG),结果显示,在未治疗和抗 PD-1 治疗的数据集中,与小肿瘤或早期肿瘤相比,大肿瘤的 CD8+ T 细胞中多种代谢通路发生显著富集,其中糖酵解/糖异生是富集程度比较高的通路(图1c-d)。DEG分析显示,来自较大肿瘤的 CD8+ T 细胞中糖酵解基因表达增加,表明代谢活跃但无效的抗肿瘤反应(图1e)。代谢与 T 细胞表型和功能相关,因此他们通过使用表达 Ova 257-264 的肿瘤细胞共培养系统进行shRNA 筛选,来探究单个糖酵解基因对 T 细胞分化的影响,Ova 257-264是肺癌模型中常用的一种明确抗原,OT-I 细胞发生激活(图1f),随后与 HKP1-ova-GFP 细胞共培养。共培养的 T 细胞最初表达效应细胞因子和颗粒酶 B (GzmB),具有适度的 Tox 和检查点蛋白表达以及表达 TCF1 的亚群。随着时间的推移,T 细胞获得效应蛋白和 TCF1 表达降低,Tox 和检查点蛋白表达升高。利用这种共培养系统,他们进行了针对差异表达糖酵解基因的 shRNA 筛选(图1c)。与已知的糖酵解最佳效应活性一致,大多数酶的敲低导致效应器功能减退状态(图1g)。丙酮酸激酶M基因 (PKM) 的敲低导致 TCF1 和 SlamF6 的上调,这是祖细胞样状态的标志(图 1g)。总之,这些结果表明干扰丙酮酸激酶后分化发生了独特的改变。
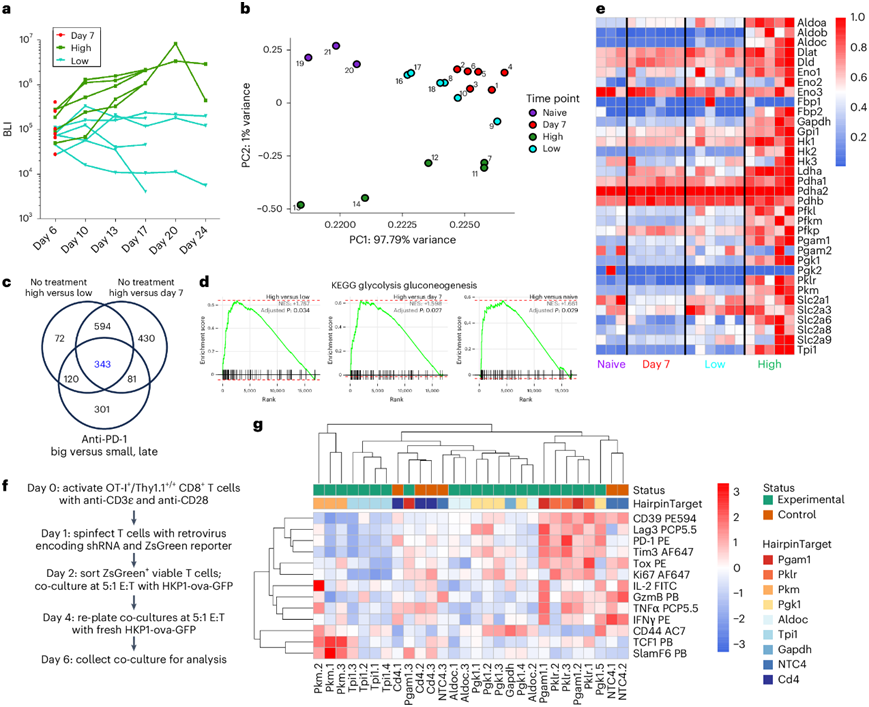
图1. 针对糖酵解酶的基因筛选确定 PKM 是 T 细胞分化的潜在调节因子。
(a) 小鼠肿瘤生长动力学的生物发光成像 (BLI) 图。(b) 主成分分析(PCA)。(c) 维恩图显示该数据集中的肿瘤表型之间的 DEG 重叠。(d) 糖酵解基因特征的差异。(e) 热图显示从 KEGG 糖酵解/糖异生数据集中选定基因在各组之间的标准化表达。(f) 针对来自(e) 的差异表达糖酵解酶的 shRNA 筛选示意图。(g) 通过平均荧光强度 (MFI) 测量的与T 细胞分化和效应功能相关的细胞因子、效应蛋白、转录因子和表面标志的标准化蛋白质表达热图。
02
PKM2 缺失影响效应 CD8+ T 细胞体外表型
PKM 有两种亚型,PKM1 和 PKM2,它们的不同之处在于第九个外显子。HKP1模型中的CD8 + TIL中的亚型分析(图1a)显示更高的 PKM2 转录本丰度。共培养的 T 细胞也显示 PKM2 表达增加且持续,而 PKM1 表达保持不变(图2a)。HKP1-ova-GFP 肿瘤模型中的幼稚或体外活化的 T 细胞的 ACT 导致 PKM2 表达高于基线水平(图2b-c),这些数据表明 PKM2 在激活时上调。
接下来,他们使用共培养系统来确定 PKM 缺乏对 T 细胞表型的影响,用 shRNA 感染活化的 T 细胞,并将它们与 HKP1-ova-GFP 细胞共培养。在初次刺激后第 6 天,shPKM T 细胞显示 SlamF6 和 TCF1 表达增加,GzmB、TIM3 和 CD39 表达降低(图2d),他们观察到 PKM 敲低后产生细胞因子的群体减少,而 TCF1high群体的丰度增加(图2e-g)。共培养的PKM2 敲除 (PKM2KO) T 细胞显示 SlamF6 和 TCF1 升高,GzmB、IFNγ 和 TNFα 降低(图2h-k)。PKM2 敲除导致 TCF1high细胞群丰度增加,而产生细胞因子的细胞群减少(图2i-k)。PKM2 缺失导致细胞活力损失极小,但增殖减少。PKM2 KO导致 CD44+ CD62L +细胞比例增加,CD44+ CD62L−细胞活力相似但略低,并且两个亚群的增殖均显著减少。PKM2 缺失对改变分化具有持久的影响,在初始刺激后第 9 天,TCF1high细胞比例持续升高,而 PKM2 野生型(PKM2WT)T 细胞具有更深的耗竭表型。他们还用不同的抗原-TCR 检测了 PKM2 缺失的影响。pmel-1 T 细胞中 PKM2 的缺失同样导致 TCF1 +、CD62L +和 SlamF6 +群体比例增加。总之,这些数据表明 T 细胞活化后 PKM2 的缺失会降低效应细胞分化。
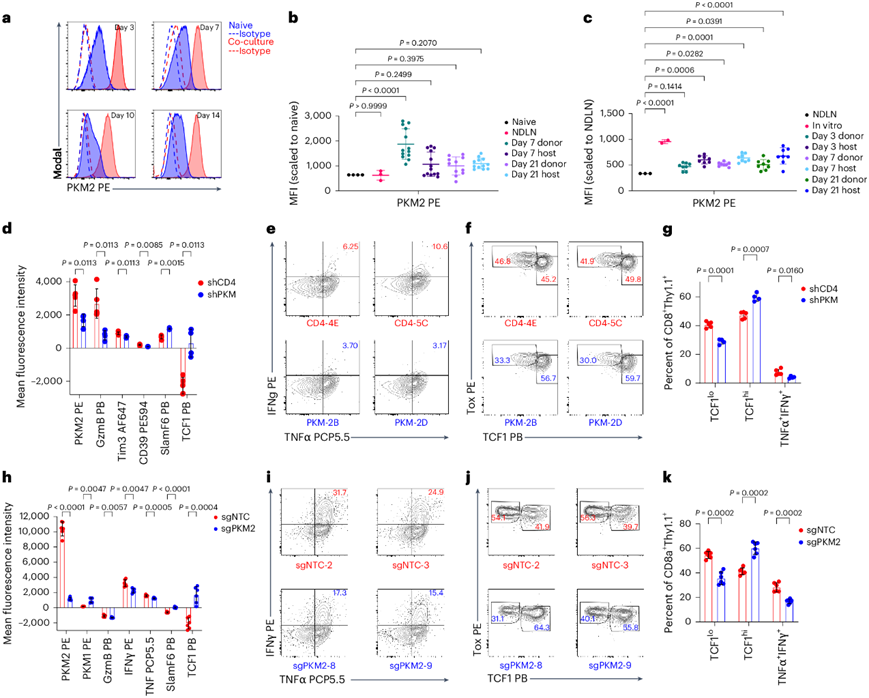
图2. PKM2 在体外和体内 T 细胞活化时上调,其缺失会导致效应器分化表型降低。
(a) 荧光直方图。(b-c) HKP1-ova-GFP荷瘤C57Bl/6小鼠T细胞PKM2表达的MFI。(d-k)流式细胞分析。
03
PKM2 缺失改变体内CD8+ T 细胞分化状态
接下来,他们评估了 PKM2KO在体内的效果。为了确保 ACT 后细胞的稳健植入,他们使用低剂量辐射(5 Gy)对淋巴细胞清除的 HKP1-ova-GFP 荷瘤小鼠进行照射。为了控制小鼠间的异质性,他们将混合的 PKM2 KO和PKM2 WT OT-I + T 细胞(以 Thy1.1 接合性为特征)过继共转移(co-ACT)到淋巴细胞清除的 HKP1-ova-GFP 小鼠中(图3a-b),并在多个时间点检测肿瘤和引流淋巴结 (dLN) 中的 T 细胞。PKM2 WT细胞迅速成为肿瘤中的主要转移群体,但仅在实验后期才显著超过 dLN 中的PKM2 KO细胞(图3c-e)。与 PKM2 WT相比,PKM2 KO细胞在重新刺激后均显示肿瘤和 dLN 中的 IFNγ 和 TNFα 表达降低(图3f-g),并且在肿瘤和 dLN 中均富集 CD44 + CD62L +细胞(图3h-i)。此外,肿瘤和 dLN 中的 PKM2 KO细胞显示 TCF1 和 Eomes 增加,而 Tbet 表达减少(图3j-m)。总之,这些表明存在伴有 PKM2 丢失的祖细胞耗竭样表型。
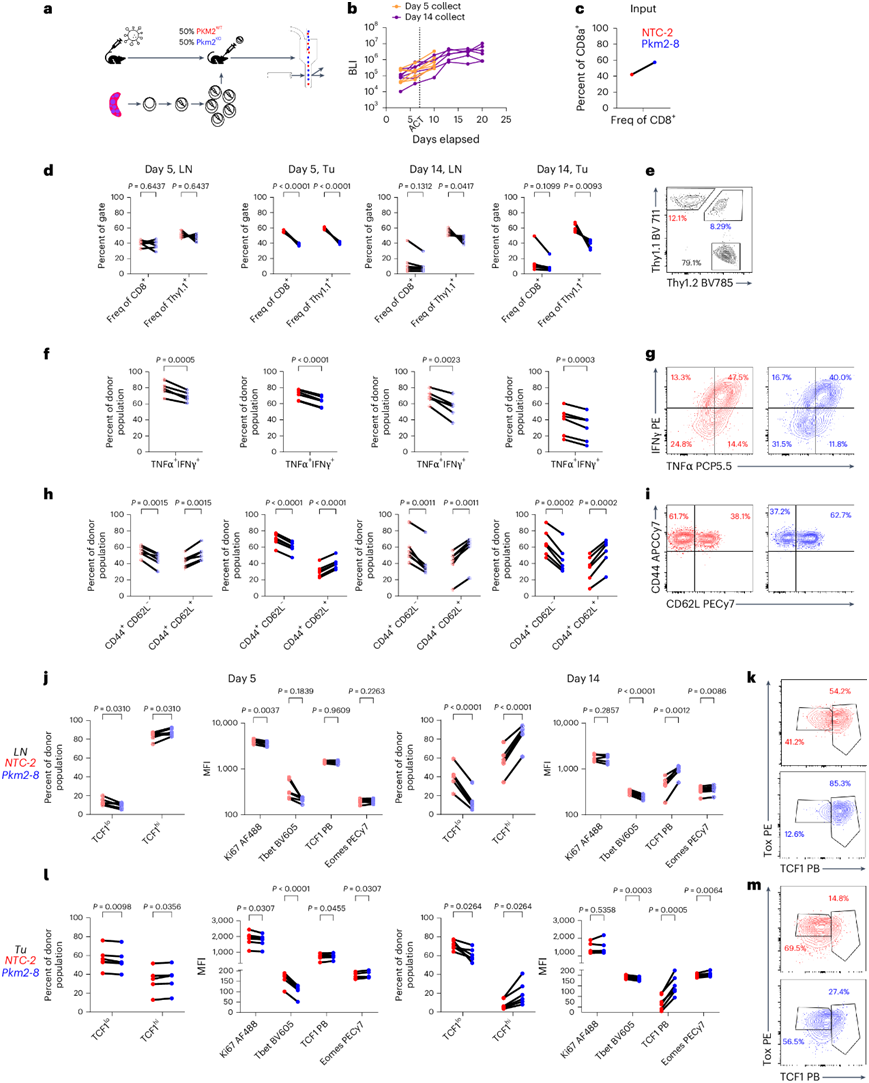
图3. PKM2 的缺失导致 NSCLC 中的 CD8 + T 细胞出现祖细胞耗竭样表型。
(a)实验方案。(b)小鼠的肿瘤负荷。(c) CD8+ T 细胞的比例。(d-i) CD8+ T 细胞群的定量。(j-m) 定量差异TCF1表达的细胞群和转录因子MFI。
04
PKM2 的缺失导致黑色素瘤 CD8+ T 细胞出现祖细胞耗竭样表型
为了确定 PKM2 在 T 细胞中缺失的影响在不同肿瘤类型中有何不同,他们采用了 B16F10-ova-GFP 黑色素瘤模型。幼稚和体外活化的 T 细胞的 ACT 都导致 PKM2 表达增加,但随着时间的推移而消退(图4a-b)。淋巴细胞耗竭的 B16F10-ova-GFP 小鼠的 Co-ACT(图4c-d)显示,肿瘤和 dLN 中 PKM2WT T 细胞占主导地位,细胞因子表达更高(图4e-i),而 PKM2KO T 细胞表现出祖细胞耗竭样表型的富集,两个组织中两个时间点的CD44+ CD62L +比例均升高,在较晚的时间点上TCF1和Eomes表达增强(图4j-o)。总之,数据表明,PKM2 在多种肿瘤类型中具有调节 T 细胞分化的一致作用,但其作用程度在不同癌症中存在差异。
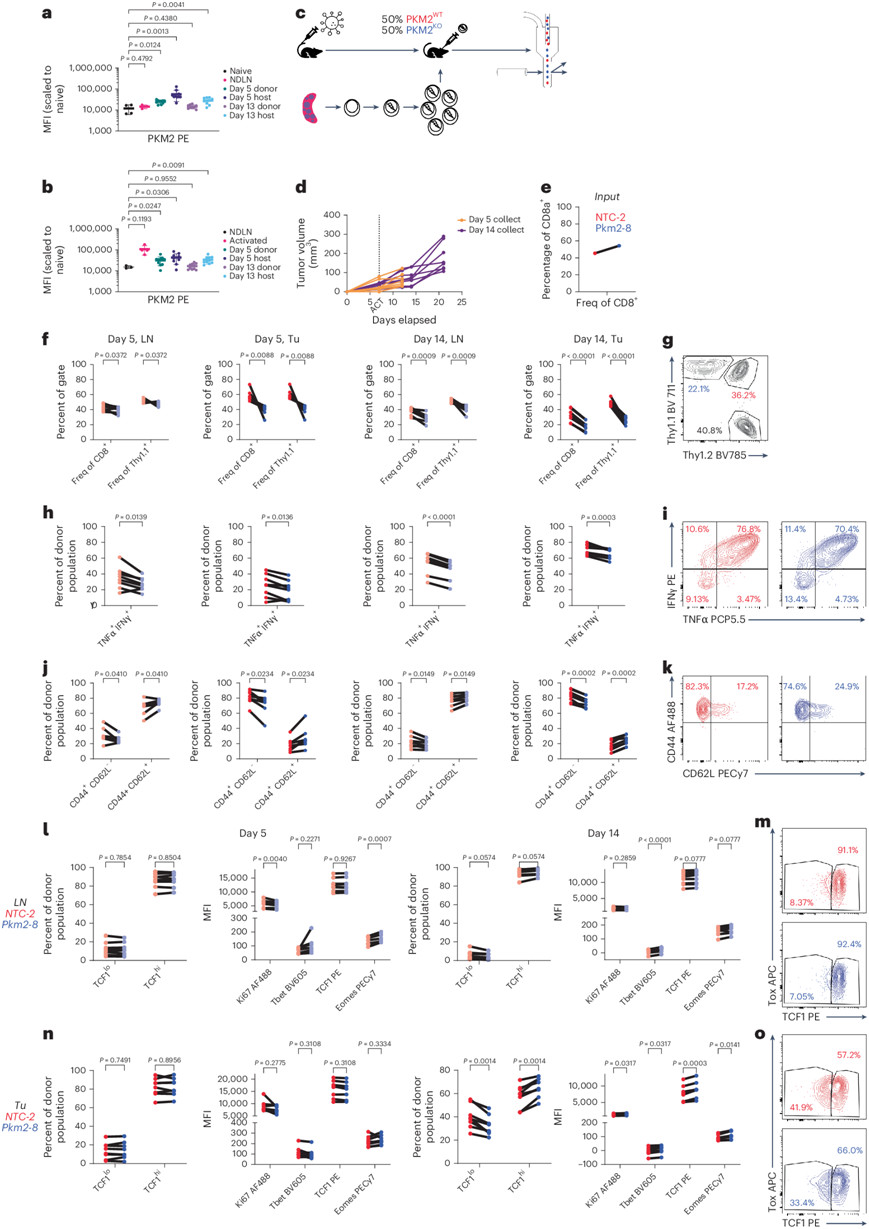
图4. PKM2 的缺失导致黑色素瘤 CD8+ T 细胞出现祖细胞耗竭样表型。
(a-b) 幼稚 T 细胞 PKM2 表达。(c)实验示意图。(d) 小鼠的肿瘤分析。(e) CD8+ T 细胞比例。(f,h,j) Thy1.1+细胞、TNFα+ IFNγ+比例以及 CD44+ CD62L−和 CD44+ CD62L+比例。(g,i,k) 肿瘤样本的代表性轮廓图。(l,n) 对具有差异 TCF1 表达和转录因子 MFI 的群体进行定量。(m,o) Tox 和 TCF1 染色代表性轮廓图。
05
PKM2KO T 细胞的 ACT 与检查点阻断产生协同作用
CD8+ T 细胞中 PKM2 的缺失导致了 TCF1high祖细胞耗竭样表型,但不足以影响肿瘤生长或小鼠整体存活率(图5a-c)。TCF1+ CD8 + T 细胞可介导 PD-1 检查点阻断的功效。为了确定 PKM2 缺乏导致的 TCF1highCD8 + T 细胞比例增加是否影响 PD-1 抑制的抗肿瘤活性,他们在 HKP1-ova-GFP 小鼠中检测了 PKM2 WT 和 PKM2 KO OT-I + T 细胞的 ACT 和抗 PD-1 处理的组合。PKM2 KO CD8 + T细胞与抗PD-1联合使用可显著抑制肿瘤生长并提高存活率(图5d-f)。在开始抗 PD-1 治疗后 4-5 天观察到 ACT 和抗 PD-1 联合处理显著控制肿瘤。PKM2 WT和 PKM2 KO T 细胞在 dLN 和肿瘤中表现出相似的丰度,其中 TCF1high细胞在 dLN 中占主导地位,而抗 PD-1处理后 TCF1low细胞富集,但肿瘤间差异很大。
为了进一步研究 PD-1 抑制的效果,他们进行了混合基因型co-ACT,然后进行抗 PD-1 或 IgG,随后进行流式细胞分析。与 PKM2WT T 细胞相比,在 PKM2KO T 细胞中,他们观察到(1)细胞比例降低;(2)祖细胞样(CD44 + CD62L +、CD127 + Klrg1 −)群增加和效应细胞样(CD44 + CD62L −、CD127 − Klrg1 +)群减少;(3) TCF1high细胞比例增加;(4) IFNγ+ TNFα+比例降低。无论基因型如何,TCF1high细胞均显示出与 TCF1low细胞相比增加的 Ki67 表达,表明增殖更多。抗 PD-1 处理导致基因型之间相似的表型:随着时间的推移,肿瘤中 CD44+ CD62L −细胞的比例增加,加速向肿瘤中 TCF1low表型的进展,并且在最新时间点肿瘤中 TCF1high细胞中 Ki67 表达更多的趋势。值得注意的是,即使使用抗 PD-1处理,对检查点阻断反应至关重要的祖细胞样群体在PKM2KO下也能更好地维持。
为了进一步探索 PD-1 抑制如何对 PKM2 KO CD8+ T 细胞产生不同的影响,他们在 HKP1-ova-GFP 模型中对来自混合基因型co-ACT 与 IgG 或抗 PD-1 结合的分选供体 TIL 进行了 RNA-seq(图5g)。对样品根据基因型、治疗状态和时间进行聚类(图5h)。PKM2KO细胞显示出记忆和祖细胞耗竭特征的富集,而 PKM2WT细胞中效应子和终末耗竭特征则相应富集(图5i-k)。Hallmark数据集显示 PKM2 WT细胞表现出更具增殖性的表型,这与其丰度成比例增加相一致,并且补体特征增强(图5l)。基因型特征差异随时间推移而变化:PKM2WT细胞早期显示出与细胞增殖和糖酵解相关的特征的富集,后期显示出与炎症相关的特征。与 IgG 相比,抗 PD-1处理富集了更具增殖性和代谢活跃的状态(图5m);这些趋势也随时间推移而变化,早期增殖相关特征存在差异,后期炎症相关特征存在差异。最后,他们研究了 PKM2 WT和 PKM2 KO细胞对抗 PD-1 治疗的不同反应。抗 PD-1 反应的子集在基因型之间是保守的,包括激活标志性特征 E2F 靶点、G2M 检查点和糖酵解等(补充表7)。不同的是,在 PKM2 WT细胞中,抗PD-1 治疗诱导与增殖相关的标志性通路富集,包括顶端连接和有丝分裂纺锤体,而在 PKM2 KO细胞中,治疗诱导氧化磷酸化 (OXPHOS) 富集,表明代谢发生了变化(图5n)。GO分析证实了这一点,在PKM2 KO细胞中ATP合成偶联电子传递、线粒体呼吸链复合物组装、OXPHOS、电子传递链等通路显著富集。OXPHOS和脂肪酸氧化都与记忆细胞能量利用有关,验证PKM2 KO细胞的祖细胞表型,并表明对抗PD-1疗法的代谢反应。总之,这些数据证实了PKM2缺失引起的分化状态改变,并证明了其对检查点阻断反应的影响。
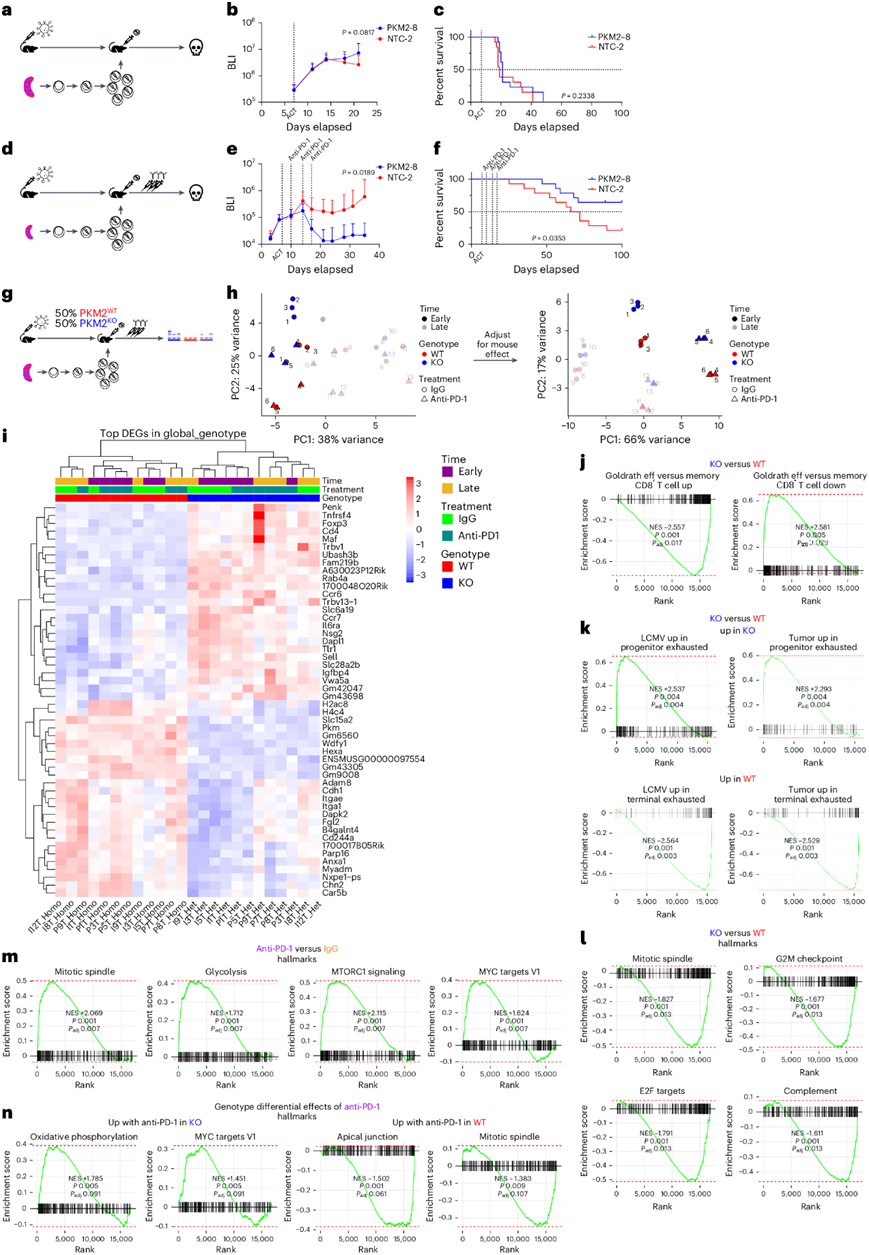
图5. PKM2 缺失会产生具有祖细胞特征的 T 细胞,从而增强 PD-1 检查点阻断的功效。
(a,d,g)实验示意图。(b,e,c,f) BLI 测量肿瘤负荷和相应的小鼠整体生存率监测。(h)主成分分析。(i) 热图显示基于供体 T 细胞基因型的前 25 个上调和下调基因的标准化表达。(j-n) 不同细胞的标志特征。
06
PKM2 KO可提高 PPP 活性
PKM2 是一种限速糖酵解酶,催化磷酸烯醇式丙酮酸 (PEP) 转化为丙酮酸。此外,PKM2 可以作为转录调节因子,改变 HIF1α、mTOR、STAT1、STAT3、STAT5 和 TGFβ/Smad2/3 信号传导的活性。他们检测了活化的 CD8 + T 细胞中 PKM2 的亚细胞定位,观察到细胞质表达,而几乎没有核 PKM2 表达,这表明 PKM2 主要在 CD8 + T 细胞中起代谢作用。然后,他们从共培养物中分选出 PKM2 WT或 PKM2 KO OT-I + T 细胞并评估糖酵解活性。与 PKM2 WT细胞相比,PKM2 KO T 细胞的糖酵解和糖酵解能力降低(图6a-b);然而,不同基因型之间的脂肪酸和谷氨酰胺氧化相似。随后,他们在多个时间点进行了稳态极性代谢物分析,以评估 PKM2 缺失引起的代谢改变。与 PKM2WT相比,从共培养中分选出的 PKM2 KO T 细胞显示糖酵解中间体的积累。PEP(可以干扰其他糖酵解反应)和 3-磷酸甘油酸都在共培养的 PKM2 KO T 细胞中在早期时间点表现出大量积累。PKM2 KO T 细胞进一步显示出磷酸戊糖途径(PPP)代谢物的富集。糖酵解和 PPP利用葡萄糖作为燃料来源,他们也共同产生几种代谢中间体,包括葡萄糖-6-磷酸、果糖-6-磷酸和甘油醛-3-磷酸。PPP 在活化的 T 细胞中生成 NADPH 和核糖-5-磷酸。值得注意的是,果糖-6-磷酸既是糖酵解中间体,也是 PPP 非氧化阶段转酮醇酶和转醛醇酶活性的重要产物,并在后期时间点富集。这些数据表明,PKM2 缺失会导致糖酵解减少和 PPP 代谢物丰度增加。
接下来,他们使用 1,2- 13C 标记的葡萄糖进行了代谢分析。使用在位置 1 和 2标记的葡萄糖可以区分由糖酵解或 PPP 活性产生的代谢物,并通过不断发展的代谢物标记模式观察多轮 PPP 活性,甚至产生大于 m+2 的电荷。他们在初次刺激后第 4 天和第 6 天从共培养中分选出转基因 T 细胞,将它们与 1,2- 13 C 标记的葡萄糖一起孵育 2 小时并进行极性代谢物分析。MetaboAnalyst分析表明PKM2 丢失对两个时间点的 PPP 都有显著影响(图6c)。在初始刺激后第 6 天,他们在 PKM2 KO T 细胞中观察到标记的 3-磷酸甘油酸和 PEP 的积累,表明糖酵解流受损,以及标记的葡萄糖酸、核糖-5-磷酸和景天庚酮糖 7-磷酸的比例增加(图6d-k)。葡萄糖酸是进入氧化 PPP 的切入点,而核糖-5-磷酸和景天庚酮糖 7-磷酸是在非氧化 PPP 中产生的两种关键产物。他们进一步观察到与单轮糖酵解或 PPP 产生的代谢物相比,同位素电荷不同的代谢物比例增加,表明 PKM2KO T 细胞中存在多个 PPP 循环(图6f-g)。在初始模拟后的第 4 天,他们观察到 PKM2 KO 对三羧酸 (TCA) 循环产生了更显著的影响,标记的 3-磷酸甘油醛、3-磷酸甘油酸、PEP、5-磷酸核糖和氧戊二酸显著增加,而 7-磷酸景天庚酮糖减少。标记模式再次显示同位素电荷,表明发生了多轮 PPP 活性。总之,这些数据表明 PKM2 缺失会导致糖酵解通量改变和 PPP 代谢物生成增加。
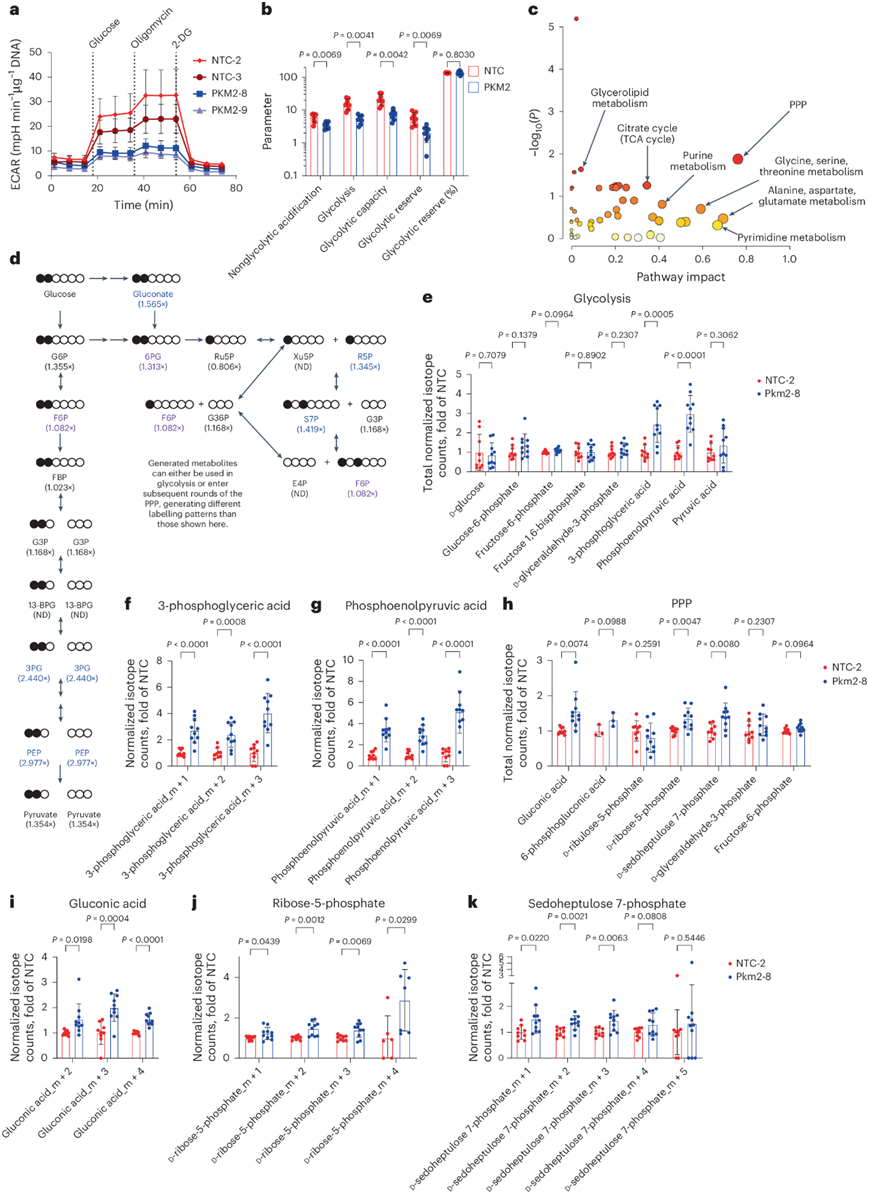
图6. T 细胞中 PKM2 缺失导致糖酵解通量减少和戊糖磷酸通路活性增加。
(a) 细胞外酸化率 (ECAR)。(b) 不同的糖酵解参数。(c) MetaboAnalyst 分析。(d) 糖酵解和 PPP 代谢物的定量。(e-k) 所有13C 标记代谢物的定量以及富集的代谢物。
07
PPP 激动剂产生独特的 TCF1 +状态
为了检测 PPP 活性升高对 T 细胞分化的影响,他们使用了 AG1,它是葡萄糖-6-磷酸脱氢酶 (G6PD)的特异性小分子激动剂,G6PD是催化 PPP 氧化期第一步和关键步骤的酶。AG1可稳定活性 G6PD 二聚体,将单体/单体界面桥接在靠近 NADP+结合位点的位置,并且在体外或体内对 G6PD 缺乏系统几乎没有影响。用 AG1 共培养处理导致 CD8 + T 细胞活力和增殖略有降低。他们进一步观察到,AG1 处理后CD44 + CD62L +细胞比例增加,AG1 处理组中 CD44 + CD62L +和 CD44 + CD62L −细胞活力相似。他们观察到 AG1 处理后 TCF1 +细胞比例增加、Eomes 表达增加、IFNγ 产生减少(图7a-d),这与 PKM2 KO表型一致。随后他们探究 PPP 激动是否通过抑制糖酵解来诱导这种表型,从而导致效应子分化受到抑制。他们从共培养中分离出 OT-I+ T 细胞,并用载体 DMSO、AG1 或己糖激酶抑制剂 2-脱氧葡萄糖 (2-DG) 处理它们 2 小时。虽然用 2-DG 进行急性处理导致糖酵解几乎完全停止,但 AG1 处理对糖酵解几乎没有影响,这表明 PPP 激动剂改变了分化,而与糖酵解损失无关(图7e)。
为了评估这些代谢过程对 CD8+ T 细胞分化的影响,他们激活细胞 1 天,然后开始用 DMSO、AG1 或 2-DG 处理。将细胞再扩增一天,然后将它们与 HKP1-ova-GFP 细胞共培养并继续处理,在初次刺激后第 6 天分选 eGFP +和 eGFP −细胞并进行RNA-seq。与 DMSO 相比,AG1 和 2-DG 处理均导致 eGFP +比例增加(图7f-g)。2-DG 处理导致 eGFP −细胞几乎完全丢失(图7f)。基于 TCF1 状态的样本之间以及基于处理的eGFP +或 eGFP −细胞之间存在较大的基因表达差异,其中 2-DG-eGFP +细胞是转录最不同的组(图7h-i)。比较 eGFP +和 eGFP −细胞发现预期的基因表达差异,包括eGFP +细胞中Sell、Fos、Id3和Tcf7增加,以及eGFP −细胞中Gzmb、Id2、Cd244a和Havcr2增加,从而确定了跨多种代谢方式保守的关键机制(图7j)。
随后,他们研究了在不同代谢刺激下 eGFP +细胞之间的基因表达如何变化。与 DMSO 相比,AG1 和 2-DG 均诱导了强烈的转录改变,而 2-DG 诱导了更广泛的差异表达(图7k)。2-DG–eGFP +和 DMSO–eGFP +样品之间的基因表达差异很大,而 AG1–eGFP +和 DMSO–eGFP +细胞之间的差异较小,在 2-DG–eGFP +与 DMSO–eGFP +比较中,药物治疗改变的基因有 9.1% 的上调基因和 25.5% 的下调基因重叠,但在 AG1–eGFP +与 DMSO–eGFP +比较中,有 53.5% 的上调基因和 73.1% 的下调基因重叠(图7k)。直接比较 AG1–eGFP +和 2-DG–eGFP +样本,证明了由 PPP 激动或葡萄糖阻断产生的TCF1+细胞中的转录异质性,与2-DG–eGFP + 样本相比,AG1–eGFP +样本中有 431 个基因表达显著增强,467 个基因表达显著降低(图7k-l)。标志分析显示,与 2-DG–eGFP +样本中的未折叠蛋白反应和缺氧相关通路相比,AG1–eGFP +样本中的增殖和炎症通路富集(图7m)。总之,这些数据表明 PPP 激动和葡萄糖阻断都会诱导 TCF1 +群体,但转录格局存在很大差异。
最后,他们探索了导致 PKM2 KO和 PKM2 WT和 AG1 和 DMSO之间基因表达差异的保守因子的上游调节因子。分析显示,在不同实验条件之间重叠的上游调节因子数量有限,包括 Bach2 和 Foxo1(图7n)。据报道,Bach2 和 Foxo1 是 TCF1 表达的上游诱导剂和记忆形成与维持的调节因子,并且在两个 RNA-seq数据集中表达均增加。他们在初始刺激后 4 天在共培养中检测了它们的表达,发现PKM2 KO和 AG1 处理均使 Foxo1 和 Bach2 的表达增加(图7o)。总之,这些数据表明 PKM2 KO和 PPP 激动之间转录因子机制的共同表达会诱导 TCF1 表达。
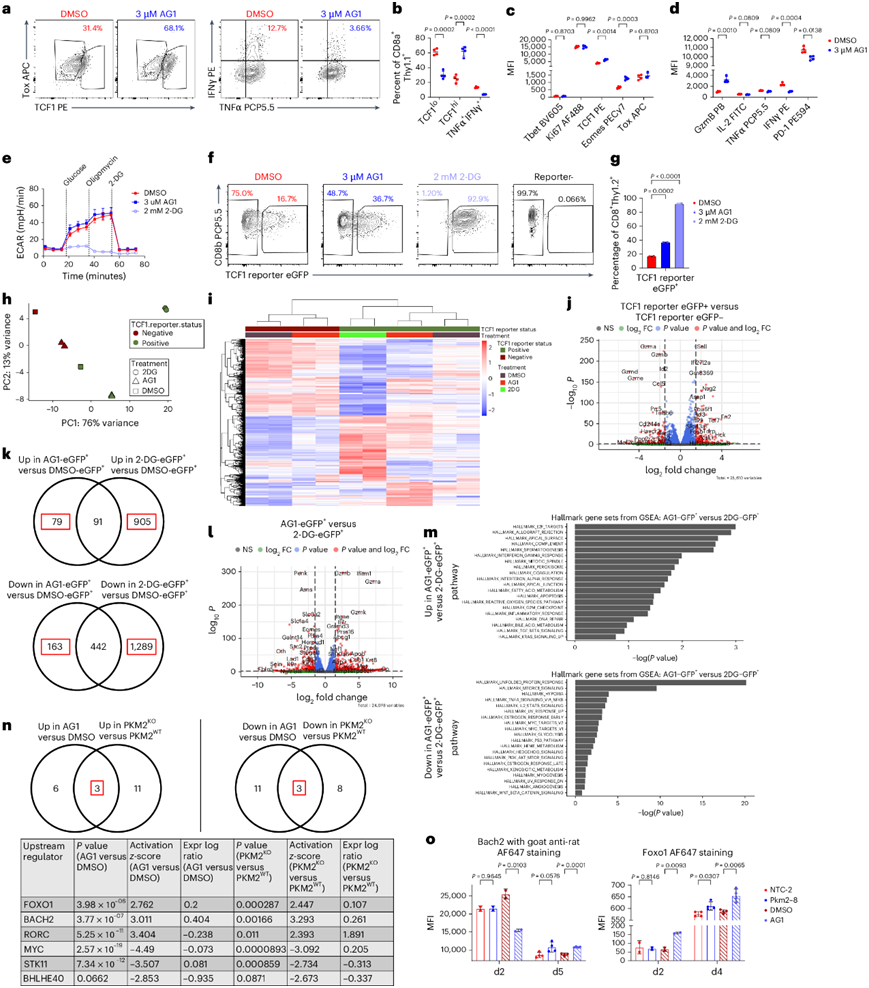
图7. T 细胞中的戊糖磷酸途径激动产生与己糖激酶阻断诱导的祖细胞表型不同的表型。
(a-b) Tox/TCF1 或 IFNγ/TNFα 的代表性轮廓图和定量。(c-d) 转录因子、效应蛋白和 PD-1的MFI。 (e) 糖酵解代谢测试。(f-g) CD8b /eGFP定量。(h)主成分分析。(i-j) 基于 eGFP 状态的前 1000 个变异基因的表达热图和比较基因表达的火山图。(k)显著差异表达基因的维恩图。(l-m) 比较AG1-eGFP+和2-DG-eGFP +样本的火山图和标志特征。(n) Ingenuity 通路分析 (IPA)。(o)流式细胞分析。
08
ACT 前的 PPP 激动与检查点阻断产生协同作用
与 PKM2KO类似,PPP 激动剂在体外导致 TCF1群体增多,因此他们检测了与抗 PD-1 联合使用是否会同样增强体内抗肿瘤功效。预处理的 OT-I + T 细胞过继转移到 HKP1-ova-GFP 小鼠中,随后小鼠接受抗 PD-1处理(图8a)。用 AG1 预处理导致 ACT 前 CD44+ CD62L+和 TCF1 +细胞比例增加(图8b-c)。AG1 预处理与抗 PD-1 联合使用可更好地控制肿瘤并提高总体生存率(图8d-f)。总之,这些结果表明 PPP 激动剂表型复制了 PKM2KO与抗 PD-1 联合使用增强的体内抗肿瘤功效。
小鼠研究表明 PPP 激动剂能够有效诱导 TCF1+祖细胞状态并与检查点阻断产生协同作用。因此,他们在人类免疫功能正常的患者来源的肿瘤类器官 (PDTO) 系统中检测了 AG1 的作用(图8g)。他们从 NSCLC 患者的标本中生成 PDTO,并在有或没有 AG1 的情况下从自体外周血单核细胞 (PBMC) 中快速扩增T 细胞(图8g)。用 AG1 处理导致 CD8+ T 细胞活力略有降低和增殖减少。检查不同的细胞亚群,观察到用 AG1 处理后CD45RA+细胞增加而 CD45RO+细胞减少。他们还观察到用 AG1 处理后所有亚群的活力降低,增殖减少,这在 CD45RO+细胞中最为明显。值得注意的是,AG1 治疗显著增加了 TCF1 表达(图8h)。AG1 处理的 T 细胞产生了更高水平的 IFNγ(图8i)。值得注意的是,在四名患者中样本中,AG1 处理的 T 细胞诱导了更高比例的凋亡类器官(图8j)。总之,这些数据表明 PPP 激动剂可能会增强人类 T 细胞的抗肿瘤能力。
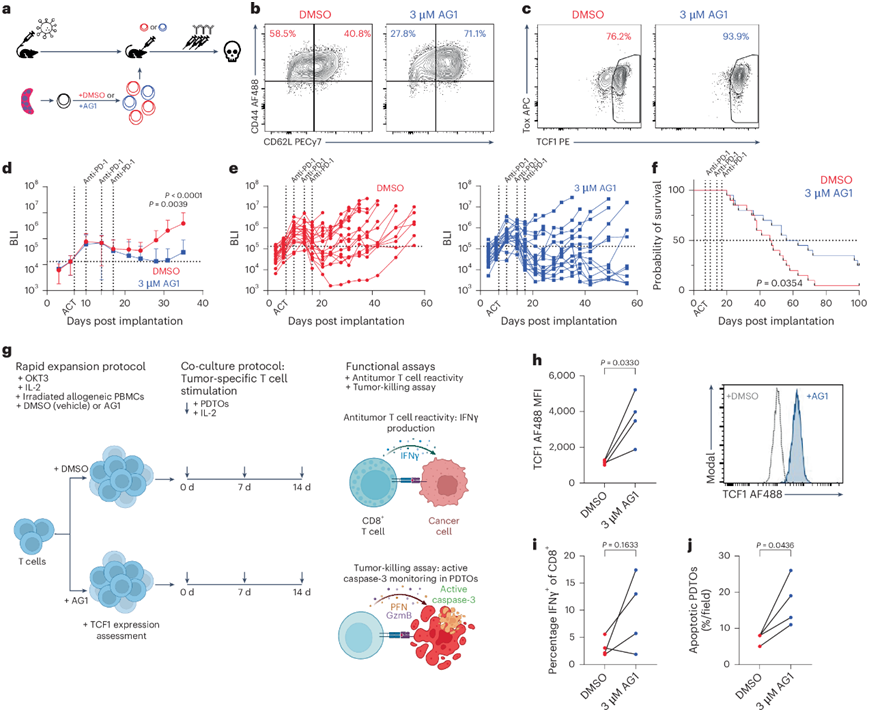
图8. 戊糖磷酸途径激动可控制小鼠和人类模型系统中的肿瘤。
(a)实验示意图。(b-c) CD44 和 CD62L 表达以及 Tox 和 TCF1 表达的流式细胞分析。(d-f) 小鼠的平均肿瘤负荷或单个小鼠的肿瘤负荷和相应的总体存活率。(g) 实验示意图。(h) 快速扩增后 TCF1 MFI 的定量。(i) 重新刺激后对 IFNγ+ CD8 + T 细胞的量化。(j) 对 12 小时内监测到的凋亡 PDTO进行定量。
+ + + + + + + + + + +
结 论
本项研究表明通过敲除PKM2靶向糖酵解会导致PPP活性升高,从而导致 TCF1high祖细胞耗竭样表型的富集和对体内 PD-1 阻断的响应性增强。PKM2 KO CD8 + T 细胞显示糖酵解通量减少、糖酵解中间体和 PPP 代谢物积累以及 PPP 循环增加,如通过 1,2- 13 C 葡萄糖碳示踪所确定的。在没有急性糖酵解障碍的情况下,PPP 的小分子激动剂激动剂处理的 CD8 + T 细胞的过继转移与 PD-1 阻断相结合增强了小鼠的肿瘤抑制,并促进了患者衍生的肿瘤类器官中的肿瘤杀灭。
+ + + + +



