

 English
English文献解读|Cancer Cell(48.8):癌周细胞毒性 T 淋巴细胞的交叉呈递削弱肝细胞癌的免疫治疗效果
✦ +
+
论文ID
原名:Pericancerous cross-presentation to cytotoxic T lymphocytes impairs immunotherapeutic efficacy in hepatocellular carcinoma
译名:癌周细胞毒性 T 淋巴细胞的交叉呈递削弱肝细胞癌的免疫治疗效果
期刊:Cancer Cell
影响因子:48.8
发表时间:2024.11.15
DOI号:10.1016/j.ccell.2024.10.012
背 景
细胞毒性T淋巴细胞(CTL)具有克隆扩增和反应性特征,是抗肿瘤免疫的关键效应细胞;它们的浸润通常是免疫检查点阻断(ICB)治疗有效的预测生物标志物。然而,只有一小部分具有富集肿瘤CTL的患者可以从ICB (抗PD-1 / PD- L1)治疗中受益,并且在接受ICB治疗后,一些患者甚至会出现疾病超进展,表明CTL在人类癌症进展中具有未曾认识的致病作用。迄今为止,CTL是否以及如何在人类癌症中发挥促肿瘤作用仍然难以捉摸。接受ICB 疗法的癌症患者可能会出现超进展性疾病,但反应性CTL 在这种情况下是否以及如何发挥致瘤作用仍不清楚。
实验设计
结 果
01

HCC 反应性 CD103 + CTL 随着疾病进展而增加
本项研究使用单细胞转录组分析 (scRNA-seq) 数据集 (GSE98638)研究肝细胞癌 (HCC)患者的肿瘤组织和配对非肿瘤肝脏和血液样本中克隆扩增的 CTL 的功能状态。在 HCC 肿瘤和血液中经常检测到具有高克隆扩增频率的 CTL,但在配对非肿瘤肝脏组织中未检测到(图 1 A )。因此,HCC 肿瘤中超过 30% 的扩增 CTL 克隆与相应血液样本中的克隆共有相同的 T 细胞受体 (TCR)。有趣的是,在肿瘤中(但在相应的血液样本中却没有)具有高克隆扩增频率的 CTL 仅表达组织驻留记忆 T (TRM) 细胞标志物ITGAE(编码 CD103)(图 1 A),并且这些细胞显示出与 TRM细胞相同的转录因子谱,表达相对高水平的AHR、PRDM1、RUNX3和ZNF683,但表达相对低水平的EOMES、TXB21、KLF2和TCF7 (图 1 B)。此外,虽然血液样本中扩增的 CTL 主要表达中枢记忆标志物CCR7和SELL,但它们在 HCC 肿瘤中的共享克隆表达相对高水平的ITGAE。因此,HCC 肿瘤中表达ITGAE的 CTL 可能源自外周免疫器官,并表现出克隆增殖和高反应性特征。与此一致,ITGAE的表达与肿瘤 CTL 中的反应性评分(包括 NeoTCR8 基因特征评分)呈正相关(图 1 C)。HCC 肿瘤中约 80% 的ITGAE hi CTL 表现出高克隆增殖(图 1 A),并显示出持续效应反应的转录特征,例如效应分子、共刺激标记、脱颗粒基因和负反馈调节受体表达增加(图 1 D)。
他们进一步使用 FACS 探测 CD103 在 48 例 HCC 相关标本(来自 23 例乙型肝炎病毒 [HBV] 相关、7 例丙型肝炎病毒 [HCV] 相关和 18 例非 HBV/HCV 相关 HCC 患者的匹配血液、非肿瘤肝脏和肿瘤组织样本)中CTL 的特异性。正如预期的那样,HCC 肿瘤中的大多数 CD103 +白细胞是典型的细胞毒性 TRM细胞,表现为 CD3 + CD8 + CD56 – CD45RO+ CD69 +细胞(图 1 E),这些细胞构成了对肿瘤裂解物而非肝脏裂解物有特异性反应的主要细胞,产生干扰素 (IFN)-γ(图 1 F),表明具有肿瘤特异性免疫反应性。与此相对应,HCC 肿瘤中大多数扩增的 TCR 克隆和大多数ITGAE hi CTL 对 HBV 不具有特异性(图 1 G),HCC 肿瘤中 CD103 + CTL的比例与血浆 HBV DNA 拷贝数无关。然而,令人难以置信的是,这些 CD103 +肿瘤反应性 CTL的比例和绝对数量在 HCC 肿瘤中,尤其是晚期 HCC 肿瘤中均有所增加(图 1 H-I)。此外,从晚期 HCC 患者中分离的 CTL 甚至表现出对肿瘤裂解物产生 IFN-γ 的能力增强(图1J)。使用 scRNA-seq 数据集(GSE98638和GSE149614),发现晚期 HCC 中的 CTL 含有更多具有高扩增频率和ITGAE表达的克隆(图 1 K-L)。总之,这些数据表明 CD103 +肿瘤反应性 CTL 与癌症进展之间存在密切联系。
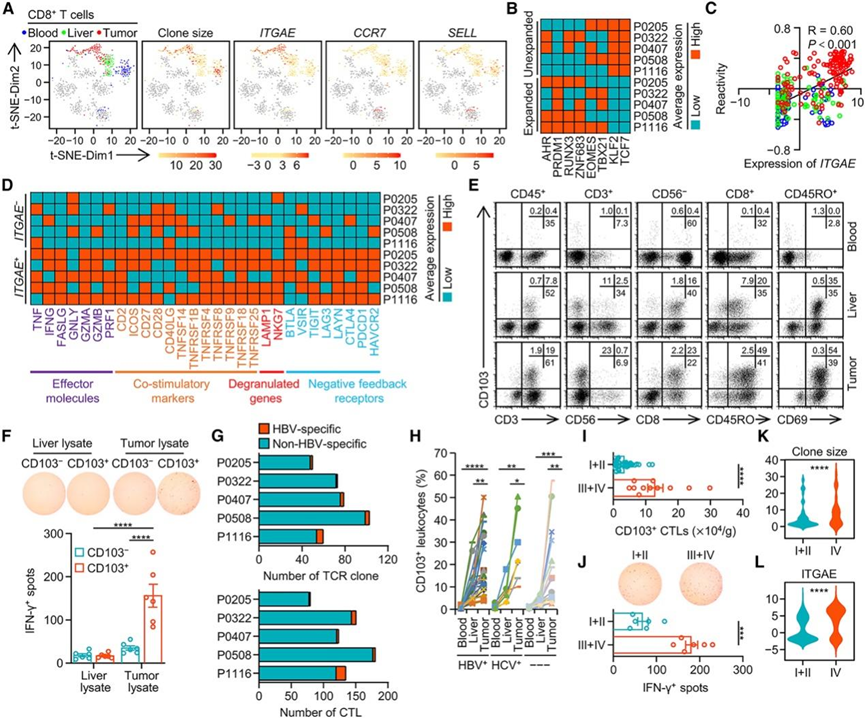
图1. HCC反应性 CD103 + CTL 随疾病进展而增加。
(A) T 细胞的 t-SNE 投影。(B) 热图显示组织驻留记忆 T 细胞 (TRM) 相关转录因子在克隆扩增和未扩增 CTL 中的平均表达。(C) ITGAE表达与 CTL 中的肿瘤反应性评分之间的相关性。(D) 热图显示ITGAE−和ITGAE+ CTL中 T 细胞相关效应分子、共刺激标志物、脱颗粒基因和负反馈受体的平均表达。(E) 对 48 名 HCC 患者的配对外周血、非肿瘤肝脏和肿瘤组织中分离的白细胞进行 FACS 分析,以检测 CD103 的表达。(F) 从 HCC 肿瘤中纯化的 CD103−和 CD103 + CTL 与 25 μg/mL自体肝脏或肿瘤块裂解物一起孵育20 小时。(G) 堆积条形图显示每个患者肿瘤 CTL 中 HBV 特异性和非 HBV 特异性细胞或 TCR 克隆的数量。(H) 23 名 HBV 相关、7 名 HCV 相关和 18 名非 HBV/HCV 相关 HCC 患者的配对外周血、非肿瘤肝脏和肿瘤组织中CD103+白细胞的百分比。(I) 48 名 HCC 患者肿瘤组织中 CD103+ CTL的绝对数量。(J) 从早期或晚期 HCC 中分离的肿瘤衍生 CTL 与 25 μg/mL自体肿瘤质量裂解物一起孵育 20 小时。(K-L) 小提琴图比较早期或晚期 HCC患者肿瘤 CTL 中的克隆大小和ITGAE表达。
02
CD103 +CTL癌周滞留预示治疗效果不佳
随后,他们评估了HCC 中CD103 + CTL的空间分布。大多数 CD103 + CTL 在 HCC 组织中积累,但不在配对的非肿瘤肝组织中积累(图 2 A)。令人惊讶的是,CD103 + CTL在肿瘤中的这种积累主要发生在癌旁区域,尤其是晚期 HCC 的区域(图 2 A-B)。相反,虽然肿瘤内 CD103 + CTL 检测到的较弱,但它们的密度与病情进展呈负相关(图 2 A)。因此,CD103 + CTL 在癌周围区域的微定位可能赋予细胞致瘤特性。癌周围 CD103 + CTL 的增加预示着早期复发,而肿瘤内 CD103 + CTL的高浸润与患者复发呈负相关(图 2 C)。单变量和多变量回归分析表明,癌周 CD103 + CTL比例是 HCC 复发的独立预测因素,具有显著的风险比,而肿瘤内 CD103 + CTL比例是良好预后的独立预测因素(图 S2 B 和 S2C)。因此,癌周 CD103 + CTL 增多且肿瘤内 CD103 + CTL 稀少的 HCC 患者的治疗结果最差(图 2 D)。
他们收集了 9 名接受抗 PD-1 治疗后接受切除术的 HCC 患者的肿瘤组织,并确定 CD103 + CTL的空间分布是否影响治疗结果。在对抗 PD-1 治疗有反应的患者的肿瘤组织中,他们检测到肿瘤内 CD103 + CTL 的密度显著增加,同时伴有癌周围 CD103 + CTL 的较低浸润(图 2E-F)。肿瘤内 CD103 + CTL 与癌周围 CD103 + CTL的比例与抗 PD-1 治疗的治疗效果呈正相关(图2G)。另一组接受抗 PD-1 治疗加经动脉灌注 (TAI) 化疗的 13 名 HCC 患者也获得了类似的结论。利用切除术后接受抗 PD-1 治疗的 8 例 HCC 组织的空间转录组学数据,证实了肿瘤内 CD103+ CTL 浸润高且肿瘤内 CD103 + CTL 与癌周 CD103 + CTL比率增加的HCC 患者对抗 PD-1 治疗有反应(图 2 H-I)。
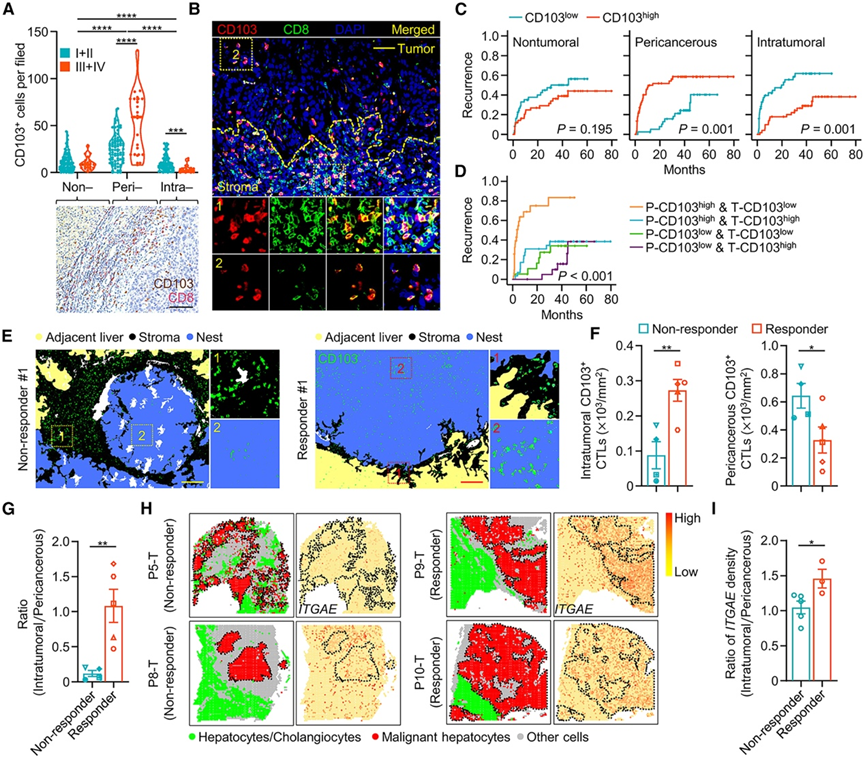
图2. 癌周 CD103 + CTL滞留预示治疗效果不佳。
(A) 免疫组织化学分析。(B) HCC 组织中 CD103 和 CD8 免疫荧光染色的代表性图像。(C-D) CD103+细胞在人肝细胞癌中的临床意义。(E-G) 肿瘤内 CD103+ CTL的空间分布与患者对 αPD-1 单药治疗的反应之间的关联。(H-I) 基于空间转录组学数据分析 8 名接受抗 PD-1 治疗的 HCC 患者切除术后的CD103+ CTL 分布。
03
DNA 高甲基化限制了 CD103+ CTL的肿瘤内积累
由于肿瘤内 CD103 + CTL 具有强效的抗肿瘤作用,他们随后探究了限制这些细胞浸润到 HCC 肿瘤中的因素。对 HCC 患者 CD8+ T 细胞中趋化因子受体转录谱的分析表明,在表达ITGAE的CTL 中CXCR3有选择性表达(图 3 A-C)。虽然表达ITGAE 的CTL也表达CCR5、CXCR4和CXCR6,但这些趋化因子受体也由不表达ITGAE的 CTL 或浸润肝脏的CD8 + T 细胞表达(图 3 A-C)。因此,CXCR3/CXCR3 配体轴代表了将 CD103 + CTL募集到肿瘤的主要信号通路。因此,CXCR3 配体CXCL9、CXCL10和CXCL11高表达的 HCC 组织表现出更高的 CD103+ CTL浸润指数(图 3 D)。但需要强调的是,与配对的非肿瘤肝组织相比,HCC 肿瘤组织中 CXCL9、 CXCL10 和 CXCL11 的表达降低(图 3 E);CXCR3 配体的这种下调可能归因于 HCC 肿瘤中相关基因的高 DNA 甲基化水平(图 3 F)。
他们进一步确定了 DNA 甲基转移酶 (DNMT) 的表达模式,并观察到与配对的非肿瘤肝脏相比,肿瘤中 DNA 甲基转移酶 1 (DNMT1) 的表达选择性增加(图 3 G)。事实上,在人肝癌细胞中过表达DNMT1不仅抑制了CXCL9、CXCL10和CXCL11的基础表达,而且还抑制了这些趋化因子在 IFN-γ 刺激下的表达。他们随后抑制了小鼠 Hepa1-6 肝癌细胞中的Dnmt1表达(图 3 H),成功上调了体内Cxcl9、Cxcl10和Cxcl11的表达(图 3 I)。Dnmt1缺陷确实增强了 CD103+ CTL 在肿瘤组织中的浸润,而这种影响可以通过阻断 CXCR3 信号消除(图 3H-J)。因此,靶向 DNA 甲基化可以恢复 CD103 + CTL的肿瘤内浸润。

图3. DNA高甲基化限制 CD103 + CTL的肿瘤内积累。
(A) t-SNE 图显示未经治疗的 HCC 患者的 CD103+ CTL的趋化因子受体谱。(B-C) 肿瘤浸润性 CD103+和 CD103 − CTL中的 CCR5、CXCR3、CXCR4 和 CXCR6 的 FACS 分析。(D) 来自 TCGA 数据集的 360 个 HCC 样本中肿瘤中CXCL9、CXCL10、CXCL11或CDH1转录水平与肿瘤 CD103+ CTL浸润指数的关联。(E) 实时聚合酶链式反应(PCR)分析9 名 HCC 患者的非肿瘤肝脏和配对肿瘤组织中CXCL9、CXCL10和CXCL11 的表达。(F) CXCL9、CXCL10和CXCL11转录水平与其甲基化状态的关联。(G) 实时 PCR 分析10 名 HCC 患者的非肿瘤肝脏和配对肿瘤组织中DNMT1、TRDMT1(编码 DNMT2)、DNMT3A、DNMT3B和DNMT3L 的表达。(H-J) 通过实时 PCR 分析小鼠肝癌组织中Cxcl9、Cxcl10和Cxcl11的表达。通过免疫组织化学分析肿瘤中 CD103+ CTL 的浸润。
04
癌周巨噬细胞专门捕获肝细胞癌中的CD103+ CTL
在癌旁区域(CD103 + CTL 的主要部位),巨噬细胞明显聚集(图 4 A),这些细胞的密度与 CD103 + CTL 的密度呈正相关。结合scRNA-seq 数据集 (GSE140228),他们确定 HCC 肿瘤中的一部分巨噬细胞构成了基质中表达CXCL9、CXCL10和CXCL11的主要细胞(图 4 B),这些细胞主要从侵袭性肿瘤边缘的肿瘤组织中分离出来(图 4 C)。在对抗 PD-1 疗法无反应的患者的肿瘤组织中,癌周巨噬细胞而非恶性肝细胞是表达CXCR3配体CXCL9、CXCL10和CXCL11的主要细胞类型(图 4 D)。
对 HCC 肿瘤的 scRNA-seq 数据集的分析(GSE140228)表明,表达 CXCR3 配体的巨噬细胞表现出高抗原呈递潜力的转录特征,这表现为 MHC I 和 II 分子以及共刺激信号的表达增加(图 4 E)。此外,他们观察到与 CD103 + CTL共同分布的癌周围巨噬细胞大量表达 HLA-DR 和 HLA-ABC(图 4 F)。相反,不能吸引 CD103 + CTL 的肿瘤内巨噬细胞仅有微弱的这些分子表达(图 4 F)。然后,他们使用 HLA-DR 和 HLA-ABC 作为标记物从 HCC 肿瘤中分离巨噬细胞。正如预期的那样,具有更多 MHC 分子的巨噬细胞分泌出更多的趋化因子 CXCL9、CXCL10 和 CXCL11(图 4 G)。使用高通量筛选 (HCS) 平台,他们证明 HLA-DRhi HLA-ABC hi巨噬细胞(而非来自肿瘤的巨噬细胞)能够快速捕获 CD103+ CTL,其特点是保留频率相对较高且细胞位移有限(图 4 H-J)。使用抗体特异性阻断 CXCR3 信号传导成功消除了HLA-DR hi HLA-ABC hi巨噬细胞诱导的CD103 + CTL 的保留(图 4 H-J)。类似地,在 transwell 系统中,当 HLA-DR hi HLA-ABC hi巨噬细胞存在于上室时,CD103+ CTL 对 CXCR3 配体混合物的迁移显著受损。此外,在具有Hepa1-6 肝癌的小鼠中,每 3 天腹膜内注射脂质体,将靶向Cxcl9、Cxcl10和Cxcl11的 siRNA 混合物特异性地递送至巨噬细胞可有效抑制这些趋化因子的表达(图 4 K),进而消除肿瘤中巨噬细胞对 CD103 + CTL 的滞留(图 4 L)。总体而言,具有高抗原呈递潜力的癌周巨噬细胞可有效捕获 HCC 中的 CD103+ CTL。
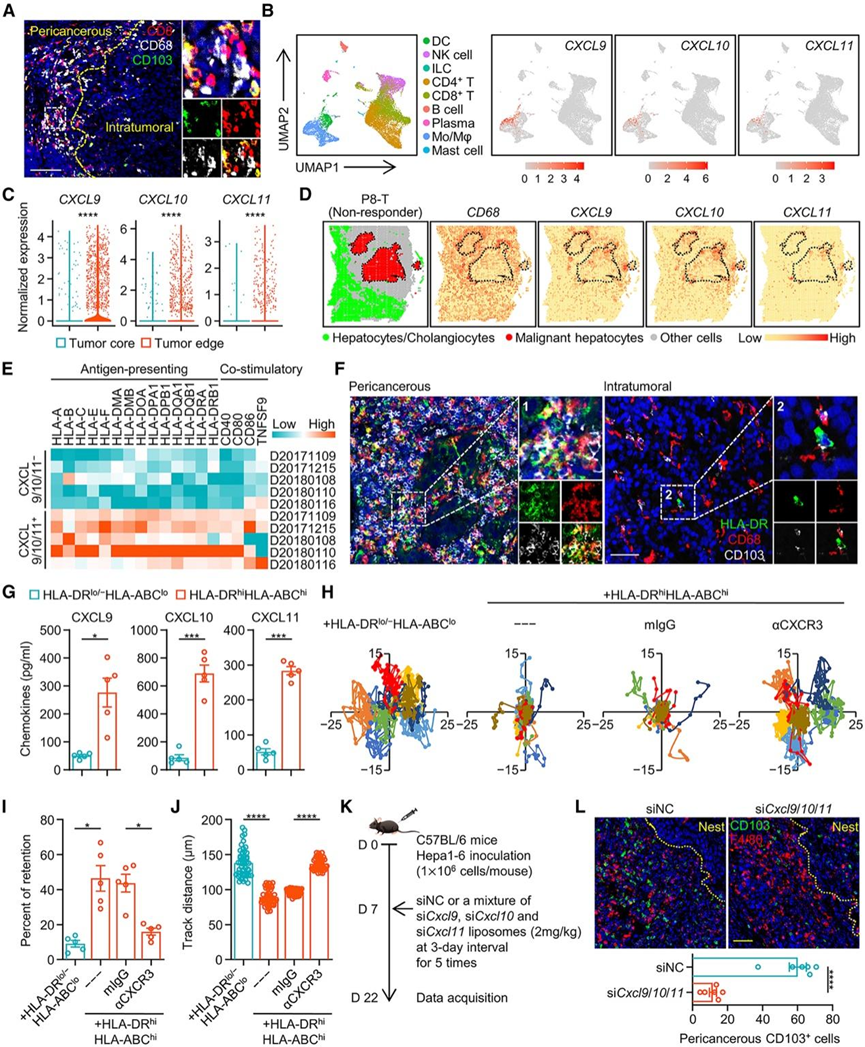
图4. 癌周巨噬细胞专门捕获肝细胞癌中的CD103+ CTL。
(A) HCC 组织中 CD103、CD8 和 CD68 免疫荧光染色的代表性图像。(B) UMAP 图显示HCC 肿瘤中不同细胞类型中CXCL9、CXCL10和CXCL11 的表达。(C) 小提琴图比较从肿瘤边缘和肿瘤核心分离的单核细胞/巨噬细胞中的CXCL9、CXCL10和CXCL11表达。(D) 基于空间转录组学数据分析ICB无反应者的CD68+巨噬细胞分布以及CXCL9,CXCL10和CXCL11的表达。(E) 热图比较CXCL9 + CXCL10 + CXCL11 +和CXCL9 − CXCL10 − CXCL11 −肿瘤巨噬细胞中抗原呈递和共刺激分子的表达。(F) HCC 组织中 CD68、HLA-DR 和 CD103 免疫荧光染色的代表性图像。(G) 通过 ELISA 检测 CXCL9、CXCL10 和 CXCL11 的分泌。(H-J)分析CD103+ CTL 的代表性轨迹、滞留百分比和轨迹距离。(K-L) 分析了癌周围 F4/80 +巨噬细胞对 CD103 + CTL。
05
巨噬细胞的交叉呈递有利于 CD103+ CTL的保留
确定了具有抗原呈递潜能的巨噬细胞有助于CD103 + CTL 在癌周的滞留后,他们评估了巨噬细胞抗原呈递对 CD103 + CTL 的影响。HCC癌周区域中共有 28.7% ± 2.9% 的 CD103 + CTL 为 Ki67 +(图 5 A);HCC 肿瘤中 CD103 + CTL的 Ki67 表达明显高于 CD103− CD8 + T 细胞。因此,在接触癌周巨噬细胞时,CD103 + CTL 可能会发生适应性激活。暴露于 HLA-DR hi HLA-ABC hi巨噬细胞后,HCC 衍生的 CD103+ CTL 获得了额外的增殖能力(图 5 B)、产生更多的 IFN-γ(图 5 C)并启动效应反应,导致表面脱颗粒标志物 CD107a 的增加,同时伴随细胞内穿孔素和颗粒酶 B 水平的降低(图 5 D)。值得注意的是,这些增殖的 CD103 + CTL 能够响应肿瘤裂解物产生 IFN-γ,但不能响应肝裂解物。这些数据表明 CD103 + CTL 和 HLA-DR hi HLA-ABC hi巨噬细胞之间存在肿瘤特异性交叉呈递。为支持这一结论,使用抗 HLA-ABC 抗体阻断 HLA-DR hi HLA-ABC hi巨噬细胞中的 MHC I 信号传导可有效抑制 CD103 + CTL的激活和效应反应,而通过抗 HLA-DR、DP、DQ 抗体阻断 MHC II 信号传导则效果不明显(图 5 E)。
值得注意的是,阻断肿瘤巨噬细胞中的 HLA-ABC 信号也会消除这些细胞捕获 CD103+ CTL 的能力(图 5 H)。同样,在患有肝癌的小鼠中,腹膜内注射脂质体以特异性地将B2m siRNA 递送至巨噬细胞,从而抑制功能性 MHC I 分子的组装(图 5 I),有效减弱了 CD103 + CTL的激活和脱颗粒(图 5 J),以及随后巨噬细胞对 CD103 + CTL 在癌周围区域的滞留(图 5 K)。因此,巨噬细胞的交叉呈递和 CD103 + CTL 在癌周围 HCC 区域的滞留是相互关联的。事实上,虽然巨噬细胞触发了 CD103 + CTL 的细胞毒性作用,但这种作用并没有导致巨噬细胞死亡,而是引发了巨噬细胞的额外激活,高表达 HLA-DR、HLA-ABC、CD80 和 CD86(图 5 L)。在与 CD103 + CTL一起培养的巨噬细胞中,上调最多的基因与 IFN-γ 相关信号有关;趋化因子 CXCL9、CXCL10 和 CXCL11 发生选择性上调(图 5 M)。相应地,阻断 IFN-γR1 会消除肿瘤巨噬细胞分泌 CXCL9、CXCL10 和 CXCL11 以及捕获 CD103 + CTL 的能力,其水平与用抗 HLA-ABC 抗体处理的细胞相当(图 5 M-N)。
值得注意的是,用于交叉呈递的肽抗原主要在溶酶体或蛋白酶体中处理。虽然在表达 CXCR3 配体的巨噬细胞中与溶酶体和蛋白酶体元素相关的基因均显著上调(图 S5 O),但抑制巨噬细胞中的蛋白酶体活性成功消除了 CD103+ CTL 的后续激活和效应反应,揭示了癌周围巨噬细胞通过蛋白酶体依赖的胞浆途径来处理肽抗原,这些肽抗原在内质网中组装到 MHC I 复合物上。事实上,表达 CXCR3 配体的巨噬细胞确实表现出编码 ER 相关降解 (ERAD) 机制的基因表达上调(图 5 O),该机制负责肽抗原转运,并且肿瘤巨噬细胞预先暴露于 ERAD 抑制剂 eeyarestatin I (EeyI) 明显削弱了其激活和捕获 CD103 + CTL的能力(图5P-S)。因此,巨噬细胞通过 ERAD 介导的胞浆途径进行交叉呈递导致 CD103 + CTL 的滞留。
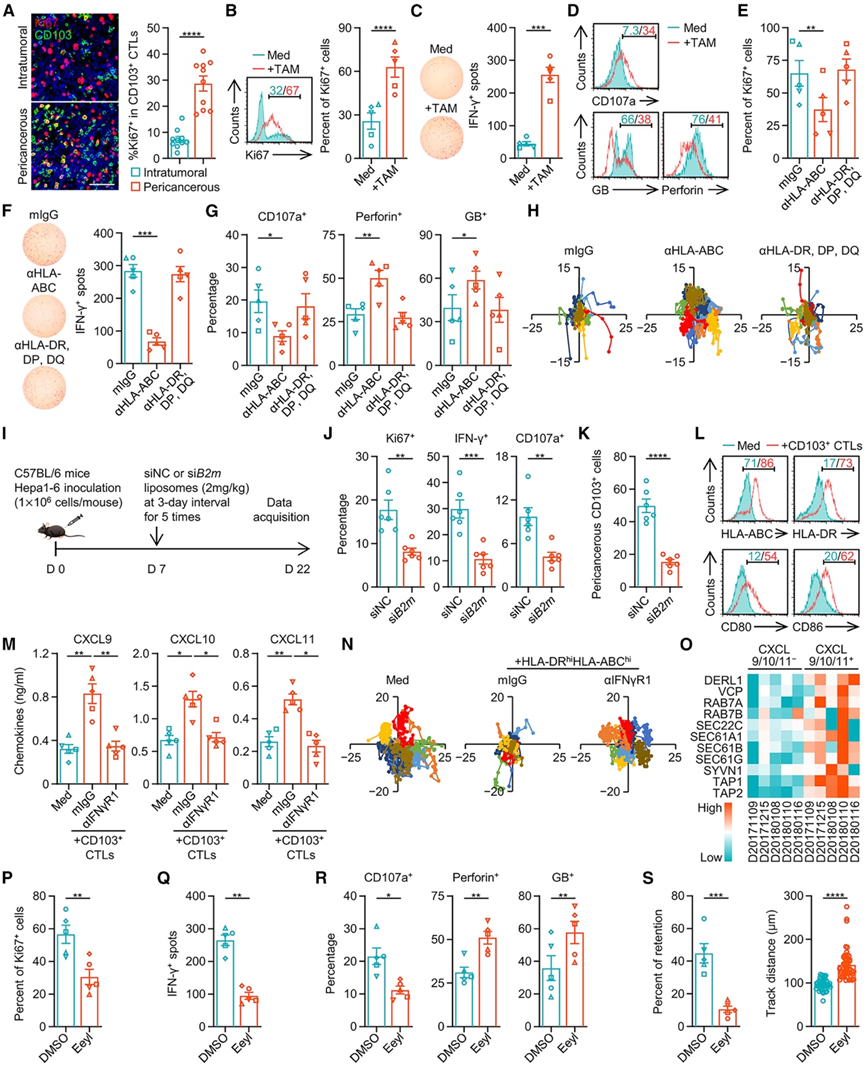
图5. 巨噬细胞的交叉呈递促进了 CD103+ CTL的保留。
(A) 肝细胞癌组织中 CD103 和 Ki67 的共聚焦显微镜分析。(B-D) 肿瘤 HLA-DRhi HLA-ABC hi巨噬细胞对 CD103+ CTL增殖、IFN-γ 产生和脱颗粒的影响。 (E-H) 分析了CD103 + CTL 的增殖、IFN-γ 产生、脱颗粒和代表性轨迹。(I-K) 分析了肿瘤 CD103+ CTL 中 Ki67、IFN-γ 和 CD107a 的表达以及癌旁区域中CD103+ CTL 的保留。(L) CD103 + CTL 对 HLA-DR hi HLA-ABC hi巨噬细胞中 HLA-ABC、HLA-DR、CD80 和 CD86 表达的影响。(M) 通过 ELISA 测量巨噬细胞的 CXCL9、CXCL10 和 CXCL11 产生情况。(N) 分析了CD103 + CTL 的代表性轨迹。(O) 热图比较CXCL9 + CXCL10 + CXCL11 +和CXCL9 − CXCL10 − CXCL11 −肿瘤巨噬细胞中参与 ER 相关降解 (ERAD) 机制的基因表达。(P-S) 分析了CD103 + CTL 的增殖、IFN-γ 产生、脱颗粒以及滞留百分比和轨迹距离。
06
反应性 CTL 激活 NLRP3 炎症小体,启动巨噬细胞致病性
先前的结果揭示了癌周围 CD103 + CTL 的不良预后价值(图 2)。为了探究癌周围 CD103 + CTL的致病机制,他们评估了癌周围 CD103 + CTL 交叉呈递后对巨噬细胞功能的影响。有趣的是,虽然与 CD103 + CTL 一起培养的巨噬细胞经历了更强的活化(图 5 L),但这些细胞选择性地产生更多的白细胞介素 (IL)-1β,而不是 IL-6、IL-10、IL-12p70 或肿瘤坏死因子 α (TNF-α)(图 6 A),并且该过程伴随着细胞内 ASC 斑点组装以及 caspase-1 和 IL-1β 的裂解(图 6 B-C),表明巨噬细胞的交叉呈递反过来导致 NLRP3 炎症小体活化。与此相对应,在HCC癌旁区域,与CD103 + CTL接触的巨噬细胞往往表达较高水平的成熟IL-1β,而在很少检测到CD103 + CTL的肿瘤内区域,巨噬细胞几乎不表达成熟的IL-1β(图6D)。此外,使用siRNA特异性抑制NLRP3炎症小体的形成或使用caspase-1抑制剂Ac-YVAD-cmk和VX-765可有效减弱CD103 + CTL介导的IL-1β裂解和分泌(图6E-F)。通过分析 scRNA-seq 数据集(GSE149614、GSE140228和GSE125449),发现巨噬细胞中的交叉呈递指数和炎症小体复合物组装之间存在正相关性。因此,用抗 HLA-ABC 抗体或 ERAD 抑制剂 eeyarestatin I 阻断巨噬细胞和 CD103 + CTL之间的交叉呈递,也会抑制巨噬细胞中的 caspase-1 和 IL-1β 裂解(图 6 G)。更准确地说,交叉呈递过程中效应 CD103+ CTL释放的穿孔素而不是颗粒酶 B 或 IFN-γ导致 caspase-1 和 IL-1β 的裂解(图 6 H-I)。值得注意的是,CTL 将穿孔素递送至靶细胞会触发钙离子内流进入细胞溶胶。36添加钙螯合剂 BAPTA-AM 可显著抑制 IL-1β 的裂解和分泌(图6 J),而 BAPTA-AM 和穿孔素抑制剂 CMA 的联合使用并没有额外抑制 IL-1β 的产生(图 6 J)。在实验的最后 3 天内,向 Hepa1-6 肝癌小鼠腹膜内注射穿孔素抑制剂 CMA 会持续抑制肿瘤巨噬细胞中的 IL-1β 裂解。
他们进一步研究了CD103+ CTL 滞留对巨噬细胞中 IL-1β 裂解的体内影响。正如预期的那样,腹膜内注射含有靶向Cxcl9、Cxcl10和Cxcl11的 siRNA 混合物的脂质体会削弱从 Hepa1-6 肝癌中纯化的巨噬细胞中的 IL-1β 裂解(图 6 K)。为了探究 IL-1β 信号传导是否是导致癌周围 CD103+ CTL 滞留介导的 ICB 治疗无反应的原因,他们阻断了接受抗 PD-L1 治疗的肝癌携带小鼠的 IL-1β 信号传导(图 6 L)。这种治疗确实有效增强了抗 PD-L1 治疗的疗效(图 6 M)。然而,在患有肝癌的小鼠中,预先注射了含有靶向Cxcl9、Cxcl10和Cxcl11的 siRNA 混合物的脂质体以抑制 CD103 + CTL的滞留,进一步抑制 IL-1β 信号传导不会改变抗 PD-L1 疗法的疗效(图 6 M)。已充分证实 IL-1β 通过触发中性粒细胞介导的血管生成发挥促肿瘤作用。事实上,抑制巨噬细胞诱导的 CD103 + CTL 在肝癌组织中的滞留也能有效抑制侵袭边缘的中性粒细胞介导的血管生成,其抑制效果与体内中和 IL-1β 相当(图 6 N-P)。一致地,具有更多癌周围 CD103 + CTL 的HCC 组织也显示出癌周围 CD15+中性粒细胞的高浸润和侵袭性肿瘤边缘的侵袭性血管生成。
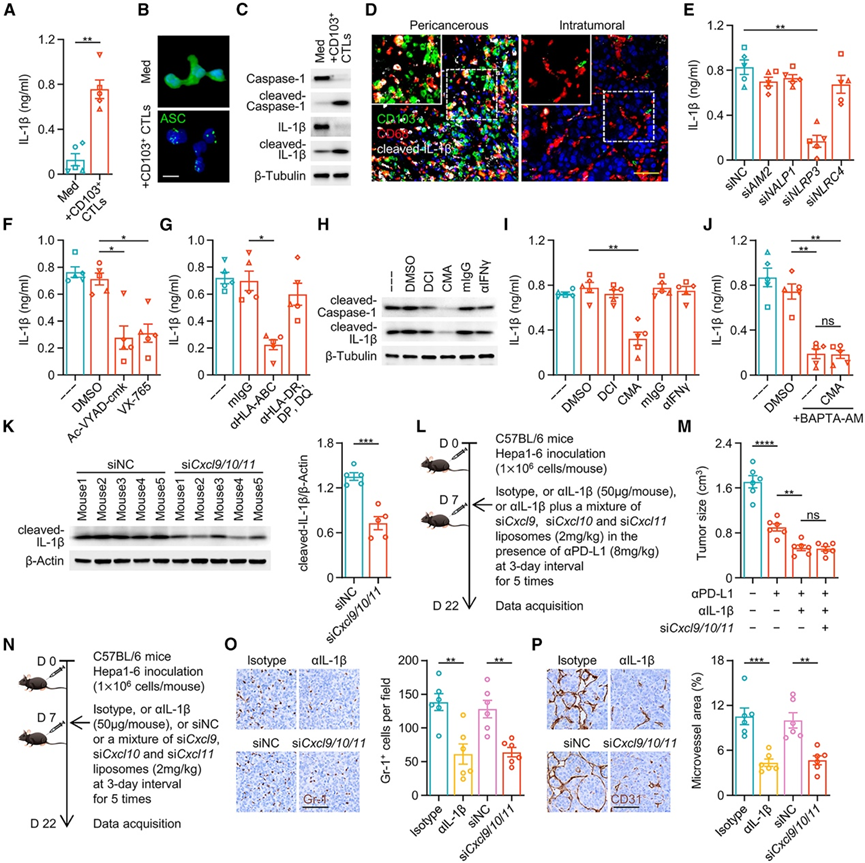
图6. 反应性 CTL 激活 NLRP3 炎症小体,启动巨噬细胞致病性。
(A-C) 肿瘤 CD103 + CTL 对肿瘤 HLA-DR hi HLA-ABC hi 巨噬细胞的 IL-1β分泌、 ASC斑点组装以及 caspase-1 和 IL-1β裂解的影响。(D) 肝细胞癌组织中 CD68、裂解 IL-1β 和 CD103 的免疫荧光染色代表性图像。(E-G) 通过 ELISA 测定 IL-1β 分泌。(H-I) IL-1β 裂解和分泌分别通过免疫印迹和 ELISA确定。(J) 通过 ELISA 检测 IL-1β 的产生。(K) Cxcl9、Cxcl10、Cxcl11敲低对小鼠肝癌巨噬细胞中 IL-1β 裂解的影响。(L-M) 分析肿瘤生长情况。(N-P) 分析小鼠肝癌侵袭边缘的中性粒细胞浸润和血管生成。
07
CD103+ CTL 的重新分布增强了免疫治疗效果
本研究的最终目标是确定抑制巨噬细胞介导的 CD103+ CTL 滞留是否会增强 DNMT1 敲低引起的 CD103+ CTL 肿瘤内浸润。于是他们注射脂质体,将靶向Cxcl9、Cxcl10和Cxcl11的 siRNA 混合物特异性地递送到具有野生型或Dnmt1 KD肝癌的小鼠的巨噬细胞中(图 7 A)。正如预期的那样,抑制巨噬细胞介导的 CD103+ CTL 滞留会触发Dnmt1 KD肝癌中肿瘤内 CD103+ CTL的额外募集(图 7 B)。同样,这两种策略的组合在抑制肝癌生长方面显示出协同作用(图 7 C)。通过注射脂质体将 siB2m特异性递送至巨噬细胞(图 7 D-E)。因此,阻断巨噬细胞介导的 CD103+ CTL 滞留为恶性细胞募集 CD103 + CTL 创造了有利条件。他们进一步研究了 CD103+ CTL的重新分布是否提高了抗 PD-L1 疗法的疗效。与 HCC 患者的治疗结果一致(图2E-I),阻断巨噬细胞介导的 CD103+ CTL 滞留加上Dnmt1KD诱导的肿瘤内 CD103+CTL 募集的联合策略可以最大限度提高抗 PD-L1 疗法在 Hepa1-6 原位肝癌模型中的疗效(图 7 F)。由于 Hepa1-6 原位肝癌模型不能完全代表对抗 PD-L1 疗法具有耐药性的系统,他们还利用了 myr-AKT/N-RasV12 驱动的自发性肝癌模型。在该模型中,DNMT1 表达显著升高,CD8+ T 细胞主要位于癌周区域,抗 PD-L1 治疗不仅未显示治疗效果,甚至还略微促进了肿瘤生长(图7G-I)。DNMT1 抑制增强了 CD8 +T 细胞浸润,并将巨噬细胞介导的 CD103 + CTL 滞留阻断与 DNMT1 抑制相结合可恢复该模型中抗 PD-L1 疗法的疗效(图 7G-I)。因此,为了改善或挽救免疫疗法的有效性,不仅要阻断恶性细胞中的表观遗传沉默,还要防止 CTL 在癌周滞留,这一点至关重要。
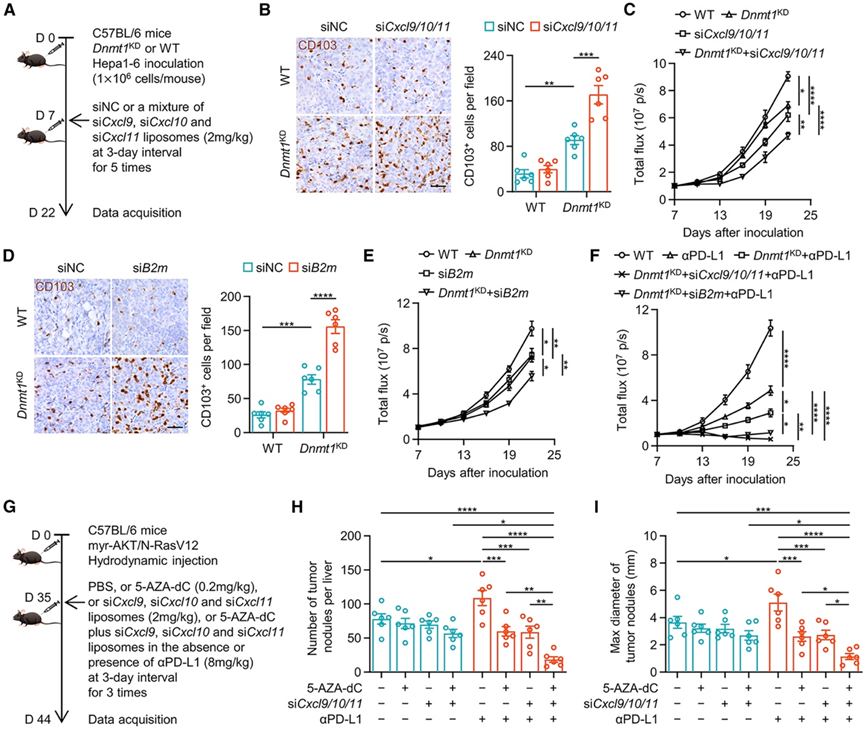
图7. CD103+ CTL 的重新分布增强了免疫治疗效果。
(A-C) 分析了肿瘤中的 CD103+ CTL 浸润和通过生物发光成像监测的肿瘤生长。(D-E) 分析了肿瘤中的 CD103+ CTL 浸润和通过生物发光成像监测的肿瘤生长。(F) 通过生物发光监测肿瘤生长。(G-I) 分析了每个肝脏的宏观肿瘤结节总数和宏观肿瘤结节的最大直径。
+ + + + + + + + + + +
结 论
癌周巨噬细胞通过内质网 (ER) 相关降解机制介导的胞质途径将抗原交叉呈递给肝细胞癌 (HCC) 中的 CD103 + CTL。该过程导致 CD103 + CTL 滞留在癌周区域,从而激活巨噬细胞中的 NLRP3 炎症小体,促进肝癌进展和对免疫疗法的抵抗。本研究对 HCC 患者进行的scRNA-seq 和空间转录组学分析表明,尽管 CD103 + CTL 具有组织驻留效应表型,但它们的聚集预示着接受多种类型治疗的 HCC 患者的临床结果不佳。相应地,重新分配 CD103 + CTL 的治疗策略可以破坏与巨噬细胞的这种致病相互作用,从而增强 ICB 治疗 HCC 的疗效。
+ + + + +



