

 English
English文献解读|Cell Host Microbe(20.6):住院早产儿肠道微生物群发育和破坏的临床后遗症
✦ +
+
论文ID
原名:Clinical sequelae of gut microbiome development and disruption in hospitalized preterm infants
译名:住院早产儿肠道微生物群发育和破坏的临床后遗症
期刊:Cell Host Microbe
影响因子:20.6
发表时间:2024.08.27
DOI号:10.1016/j.chom.2024.07.027
背 景
新生儿在分娩期间和分娩后从母亲和环境中获得身体各个部位的微生物。新生儿的肠道微生物群是免疫和营养发育所必需的器官,其不断积累微生物。微生物群的发育受多种因素影响,包括环境、分娩方式、抗生素暴露、饮食和妊娠周龄 (GA)。全世界有10.6% 的婴儿是早产儿(定义为妊娠第37 周前出生),许多婴儿需要在新生儿重症监护病房( NICU)接受数月的初始住院护理。NICU住院治疗和随之而来的必要干预措施,包括不同于家庭环境的抗生素和改变喂养干预,会极大地影响肠道微生物群的发育。与足月婴儿快速获得共生厌氧菌不同,早产儿肠道最初接种的细菌包括医院内病原菌,包括葡萄球菌、肠球菌和肠杆菌科。这些医院内病原菌在早产婴儿的粪便中大量存在,往往主导其宿主的肠道微生物群(占整个菌群的 50% 以上),并在NICU环境中的婴儿之间传播。影响病原菌和共生菌进入和排出早产微生物群的具体临床变量和暴露仍未完全了解。
实验设计
结 果
01

住院早产儿的肠道菌群组装
为了确定早产儿肠道中的细菌群落,研究团队首先对1479份粪便样本进行了宏基因组测序,这些样本来自在密苏里州圣路易斯(56 %)、俄克拉荷马州俄克拉荷马市(33 %)和肯塔基州路易斯维尔(10 %)的NICU住院的96例无坏死性小肠结肠炎(NEC)早产儿,几乎每天收集一次。这些婴儿是一个前瞻性队列的子集,该队列调查了住院期间未发生NEC的早产儿的NEC细菌病原学。
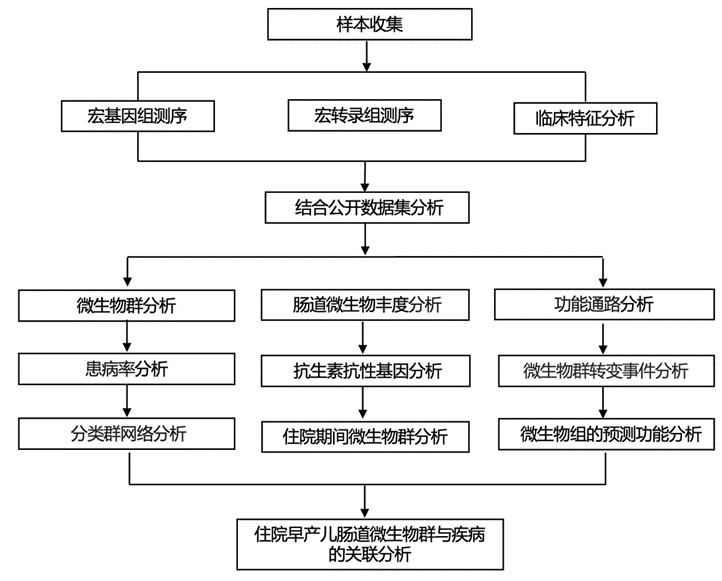
住院早产儿的肠道菌群发育遵循一种共同的模式,从最初葡萄球菌属和其它杆菌在几乎所有婴儿中占优势,到由致病菌定义的群落变化。宏基因组数据支持早产儿最早微生物组组装的这些精心设计的轨迹。本研究队列中早产儿的肠道菌群在出生后的第一个月内迅速多样化(图 1B),积累了多种分类单元(图 1C),并从表皮葡萄球菌占优势转变为由潜在致病菌定义的群落,特别是肺炎克雷伯菌(K. pneumoniae)、粪肠球菌(E. faecalis) 和大肠杆菌(E. coli)(图 1C,图S1A)。他们结合了公开的数据,表明肠杆菌科、粪肠球菌和表皮葡萄球菌等致病菌非常普遍,并且通常以 >50% 的丰度主导单个婴儿的微生物群(图S1B-C)。值得注意的是,他们还发现了假定有益的早期定植微生物,包括双歧杆菌属和韦荣球菌属,这表明即使在 NICU 住院期间也会获得重要的共生菌(图S1A-C)。
最早的早产肠道菌群受从 NICU 环境中获得的微生物的影响。为了系统地表征从环境来源获得的特定物种的微生物,他们共同组装、并分类注释了婴儿特异性宏基因组组装基因组 (MAG),总共去复制了 1176 个高质量和中等质量MAG,生成了一组 197 个物种级代表性基因组。利用该 MAG 数据库,他们使用 inStrain方法对所有宏基因组样本对的物种特异性成对平均核苷酸同一性 (ANI) 值进行了分析。
他们在所有物种和样本对的多个样本中总共发现了 28251 个相同菌株的实例,其中 26378 个 (93.4%) 来自同一婴儿在一段时间内采集的样本。在所研究的 96 名婴儿中,有 82 名 (85.4%) 与另一婴儿共有至少一种细菌物种的至少一种菌株。仅当来自同一婴儿对的≥2个独特样本共有一个菌株时才专门计算菌株共有事件,以限制假阳性关联。表皮葡萄球菌和艰难梭菌菌株是在多名无关婴儿的粪便样本中最常见的物种(图 1D-E)。他们没有观察到菌株共有事件与患病率之间的相关性。表皮葡萄球菌的菌株共有事件比具有可比跨队列流行率的其他菌种更常见(图 1 D),这表明环境因素可能不成比例地促进其在 NICU 中的传播,或者这些菌种能够很好地适应在医院环境中存活。艰难梭菌的流行更容易发生(图 1 D)。表皮葡萄球菌的流行率随着出生天数的增加而下降(图 S1C),并且与carbapenem类和糖肽类暴露呈正相关(图 1 F)。相反,艰难梭菌的流行率随着 NICU 出生天数的增加而增加,并且与氨基糖苷类、第四代头孢菌素和carbapenem类药物的给药呈负相关(图 1 F)。这些观察结果表明生物体特有的获取和传播动态,可能与不同的环境影响有关。

图1. NICU 中最早的早产儿肠道菌群定植。
(A) 研究设计。(B) 主成分分析。(C) 九个选定分类群的出生后 DOL 患病率。(D)早产儿中共有菌株分析。 (E) 两个分类群在无亲缘关系的婴儿对中发生此类事件最多的菌株共有事件的网络表示。(F) 抗生素暴露对前 80 天整个对照队列中表皮葡萄球菌和艰难梭菌患病率的影响。
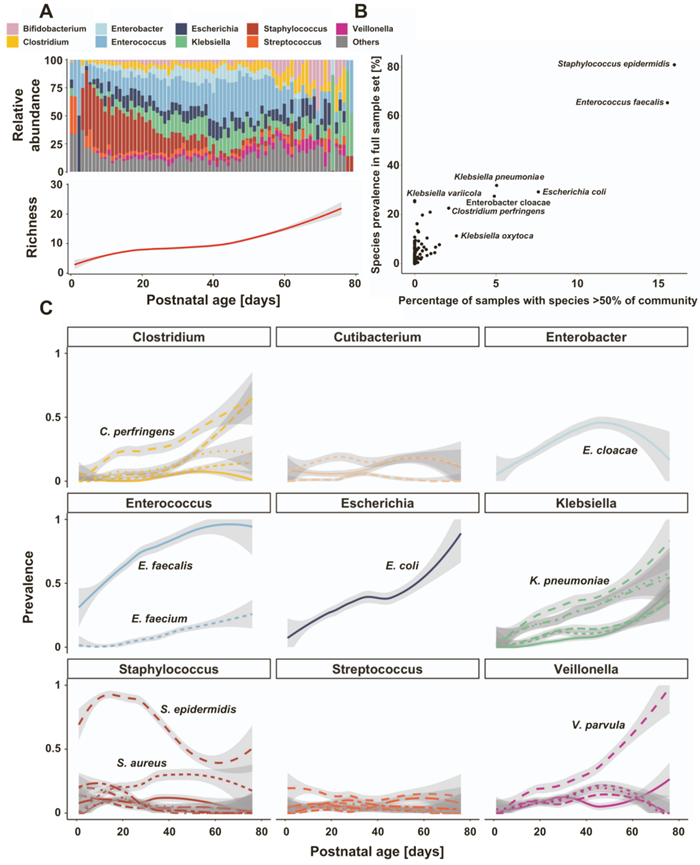
图S1. 住院期间早产儿肠道微生物群的聚集。
(A) 出生后几天平均相对微生物群丰度(上)和丰富度(下)。(B) 早产儿肠道微生物群的物种患病率和相对丰度大于50%。(C) 不同属在出生后一天的流行率。
02
微生物组转变将致病菌和抗生素耐药性引入早产儿肠道微生物组
肠道微生物组成的变化可迅速改变宿主免疫系统的微生物线索。此类微生物组转变是导致成年人群胃肠道疾病和感染(如炎症性肠病和旅行者腹泻)病理生理的原因。这些事件的特点是,两次采样之间的间隔群落变化超出了定期观察到的患者内变异所设定的界限,并且与患者间变异难以区分。
为了表征分类学微生物群转移的频率、原因和后果,他们对从同一参与者连续采集的样本中的 Bray-Curtis 差异性进行了分析。20 天内连续样本的组成比来自无关个体的样本更相似,他们发现了 131 个微生物群转移事件,定义为个体内样本间 Bray-Curtis 差异性大于个体间比较的平均差异性。转移事件发生的时间明显早于样本采集的平均出生后天数,表明接近出生时不稳定性更大,并且与分类单元的出现和消失有关。总体而言,转移表明微生物组不稳定的间断,伴随致病菌丰度的剧烈变化(图 2 A)。与无转移的样本对相比,转移更有可能将致病菌(即阴沟肠杆菌、大肠杆菌、粪肠球菌和肺炎克雷伯菌)引入早产儿的肠道(图 2 A)。转移还耗尽了假定的有益类群,包括长双歧杆菌和韦荣球菌种以及上面引入的相同致病菌。转移中引入的常见致病菌通常会在随后的 10 天内持续存在(图 2 B),这表明这些群落改变对正在发育的早产肠道微生物组具有持久的影响。
前 14 天接触抗生素,尤其是广谱药物,如第三代和第四代头孢菌素和carbapenem类以及针对革兰氏阳性菌的药物,包括糖肽类和林可酰胺类,与微生物群转变有关。值得注意的是,与非转变时间点相比,在微生物群转变期间引入了先前未在同一婴儿肠道微生物群中发现的抗生素耐药性基因(图 2 C),这些基因编码多种抗生素耐药机制(图 2 D),包括广谱 β-内酰胺酶,这表明在转变前后引入的致病菌可能由于其抗生素耐药性基因库而增殖。这些数据表明,微生物群的变化可能是临床变化的关键时刻,将抗生素或抗菌素耐药性病原体引入住院早产儿的肠道,从而进一步扰乱微生物群的发展。
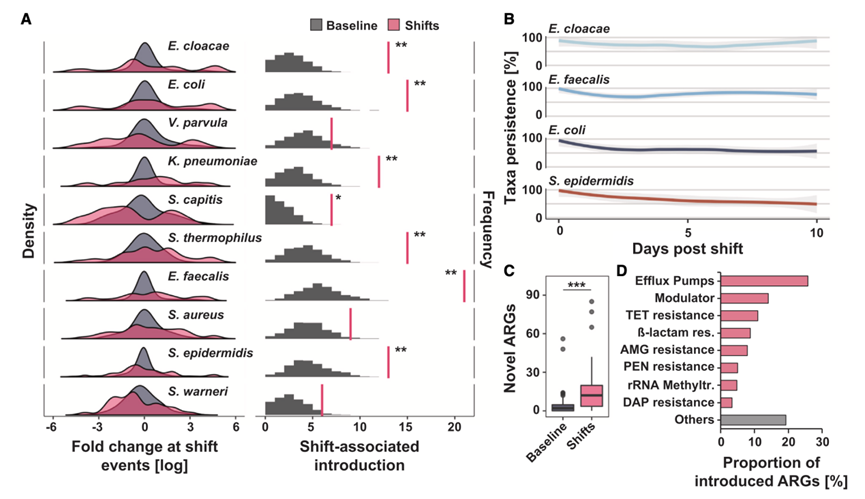
图2. 抗生素相关的微生物组变化导致早产儿肠道中微生物丰度发生变化。
(A) 转移事件相关的选定物种丰度变化。(B) 在转移事件中引入的选定分类群在转移后 10 天内的持久性。(C) 抗生素抗性基因 (ARG) 的数量。(D) ARG 分类。
03
NICU 暴露组影响住院期间微生物群的发展
除抗生素外,住院的早产儿还经常接触一系列宿主指导的药物和干预措施。这些变量和其他变量,例如出生方式和医院、母亲产前暴露和婴儿饮食,统称为暴露组,并塑造早产儿肠道微生物组轨迹。为了全面描述NICU暴露组如何影响住院期间早产儿肠道微生物群的组装和功能成熟,他们将上述96名婴儿的 1479 个宏基因组学谱与来自同一研究人群的 92 名不同早产儿的 409 次粪便的宏基因组学数据整合在一起,从该集合中,他们从出生后 88 天内(DOL:出生后的天数)收集的 188 名早产儿中合成了 1888 个分类学概况,其中包含大量临床元数据,包括基线变量、母亲信息、抗生素疗法、饮食暴露和非抗生素药物使用。由于生命最初几周内微生物组成发生了快速变化,并且某些药物对微生物有特异性作用,他们假设出生后暴露对微生物组成的个体和集体影响比产前暴露更大。为了验证这一假设,他们使用重复测量置换方差分析(重复PERMANOVA)探究了元数据变量和 NICU 暴露对肠道微生物组的 Bray-Curtis 差异性的影响(图 3 A)。正如预期的那样,样本信息解释了肠道微生物组 60% 的分类学变异和 59% 的功能变异。他们将变量分为三类:基线/母体/饮食、抗生素和非抗生素药物,发现只有 3/27(11%)的基线/母体/饮食变量与分类学变异显著(图 3 B)。出生后年龄是最具解释力的特征,解释了 4% 的分类学变异(图 3 A)和 6% 的功能变异。配方奶粉和母乳暴露的累计总量对分类学变异的贡献率为 1%。其他任何测试的基线或母体变量(包括医院地点、分娩方式和 GA)均与出生后 3 个月的微生物组分类或功能潜力无显著相关性。在后续分析中,他们关注出生后年龄,因为 GA 与宏基因组变异没有显著相关性。为了进一步确定出生后年龄对微生物组变异的影响,他们将本研究的队列分为 10 个 DOL 窗口,将 DOL > 50 的婴儿样本集中在一起。出生后年龄与所有 DOL 窗口中的微生物组发育显著相关,其中最大效应为 1.5% 的方差。由于母体和基线变量与足月婴儿的微生物组差异有关,这些数据使他们假设,对于 NICU 住院的早产儿,早期生活暴露对微生物组发育的贡献大于产前暴露。
总的来说,前 14 天内 6/11 (55%) 的抗生素类别暴露与微生物组分类组成显著相关,并贡献了 6% 的方差(图 3 B)。令人惊讶的是,样本采集前 7 天内68%的非抗生素药物暴露与分类组成有关,共同贡献了 10% 的方差(图 3 B)。由于 NICU 中的药物经常同时使用,他们确定了药物共同使用的频率,以了解非抗生素药物的影响。除了氨苄西林和庆大霉素、万古霉素和庆大霉素以及咖啡因和庆大霉素共同使用外,大多数药物暴露并不重叠。综上所述,这些发现表明,出生后最初几周内新生儿重症监护室 (NICU) 内的微生物群组装动态在更大程度上是由出生后环境暴露而不是出生前生物学决定的。
为了确定 NICU 暴露组如何塑造分类单元丰度轨迹,他们使用 MaAsLin2 建立了每个特征的广义线性混合模型 (GLMM)。他们将参与者、医院地点和研究作为随机效应,并评估了所有 62 个基线/母亲/饮食、抗生素和非抗生素药物变量(图 3 B)。该方法可以校正已知的混杂因素并反卷积同时暴露的影响,但不解决变量之间的相互作用。虽然产前和母亲变量不能解释整体分类学差异(图 3 B),但出生前母亲门诊抗菌药物和 GA 增加均与单一物种的增加有关(图 3 C)。他们假设产前和基线变量在生命早期可能更为重要,因为他们没有观察到它们对整体微生物组组成产生影响(图 3B)。他们将数据集缩减为来自前 10 个 DOL 的 73 名参与者的 190 个样本,并仅评估了 27 个基线/母体/饮食变量,同样没有发现影响分类的母体或基线变量。这些结果表明,母体和基线变量对早产住院婴儿的早期微生物组组成影响微乎其微。
出生后年龄的增加与表皮葡萄球菌、沃氏葡萄球菌和贪婪梭菌的减少有关(图 3C)。DOL 还与厌氧菌的增加有关,例如韦荣球菌属、艰难梭菌、Finegoldia magna和梭菌属,以及与潜在致病的肠杆菌科细菌有关,包括克雷伯氏菌属和大肠杆菌(图3C)。累积饮食暴露(编码为取样前接触配方奶或母乳的天数)与主要潜在病原体(如克雷伯氏菌、大肠杆菌和葡萄球菌属)的变化有关(图 3C)。总的来说,采样前 14 天内使用抗生素与 24 种细菌丰度减少和 5 种细菌丰度增加相关(图 3 D)。之前 14 天内暴露于氨基糖苷类药物是队列中最常见的暴露,影响了50%的样本,该暴露与F. magna、S. aureus和C. difficile丰度减少以及S. epidermidis丰度增加有关(图 3 D)。β-内酰胺类抗生素与肠杆菌科细菌丰度减少有关,其中第四代头孢菌素对大肠杆菌丰度减少的影响最强。青霉素(氨苄西林、替卡西林-克拉维酸、氨苄西林-舒巴坦)和第三代头孢菌素与多种克雷伯氏菌种丰度下降有关(图 3 D)。第三代头孢菌素也与粪肠球菌丰度增加有关,这可能是由于它们固有的头孢菌素耐药性。万古霉素与粪肠球菌丰度下降有关,这可能反映了韦荣球菌属菌株水平变异的易感性。韦荣球菌属的细菌丰度也因最近接触第一代头孢菌素和林可酰胺类药物而减少。因此,他们证明许多潜在病原体(肠杆菌科、葡萄球菌属和粪肠球菌)以及假定的有益分类群(包括韦荣球菌)的丰度减少。这些抗生素的共同暴露可以明显改变肠球菌和肠杆菌科的肠道微生物群的丰度。
接下来,他们研究了非抗生素药物与肠道微生物组分类内容之间的具体关联。与主要与分类减少相关的抗生素不同,非抗生素药物暴露与双向变化相关,关键分类单元增加 41 个,减少 38 个,总体患病率超过 10%(图 3 E)。有趣的是,非抗生素暴露的变化系数通常大于抗生素暴露(图3C-E),这种影响已在患有心脏代谢疾病的成年人中观察到。单独物种与非抗生素暴露显著相关。克雷伯氏菌属是受影响最频繁的属,口服多种维生素和法莫替丁与 6 种克雷伯氏菌增加有关,而芬太尼、咪达唑仑和葡萄糖酸钙与 8 种克雷伯氏菌减少有关(图 3 E)。咖啡因是总体上最常见的药物,影响了88%的样本,它与艰难梭菌减少、两种克雷伯氏菌增加以及粪肠球菌增加有关(图 3 E)。口服含铁药物影响了近 1/3 的样本,它与表皮葡萄球菌和密歇根克雷伯氏菌减少以及不规则弧菌和大型弗朗西斯菌增加有关。虽然其中一些变化可能是由于补铁年龄增加所致(接触铁剂的婴儿为 43 天,而未接触铁剂的婴儿为 22 天),但这些数据也表明,补铁可能导致有益的韦荣球菌和芬戈尔德球菌增加,反映出更成熟的微生物组组成。在接触芬太尼后,粪肠球菌、Cutibacterium avidum、F. magna、雷伯氏菌属和V. dispar的相对丰度降低有关。这些减少可能与阿片类 (opioid) 药物引起的运动能力变化有关,也可能继发于导致肠道微环境差异的其他微生物变化。粪肠球菌丰度增加通常与非抗生素药物有关,包括类固醇(地塞米松和氢化可的松)、含铁的多种维生素和胆钙化醇。有趣的是,肠球菌必须获得铁才能在肠道中发挥毒性和厌氧生长。这些结果表明,在 NICU 住院期间,早产儿肠道中的致病菌和关键肠道共生菌的分类丰度变化与抗生素和非抗生素暴露或施用这些抗生素的医疗条件有关。
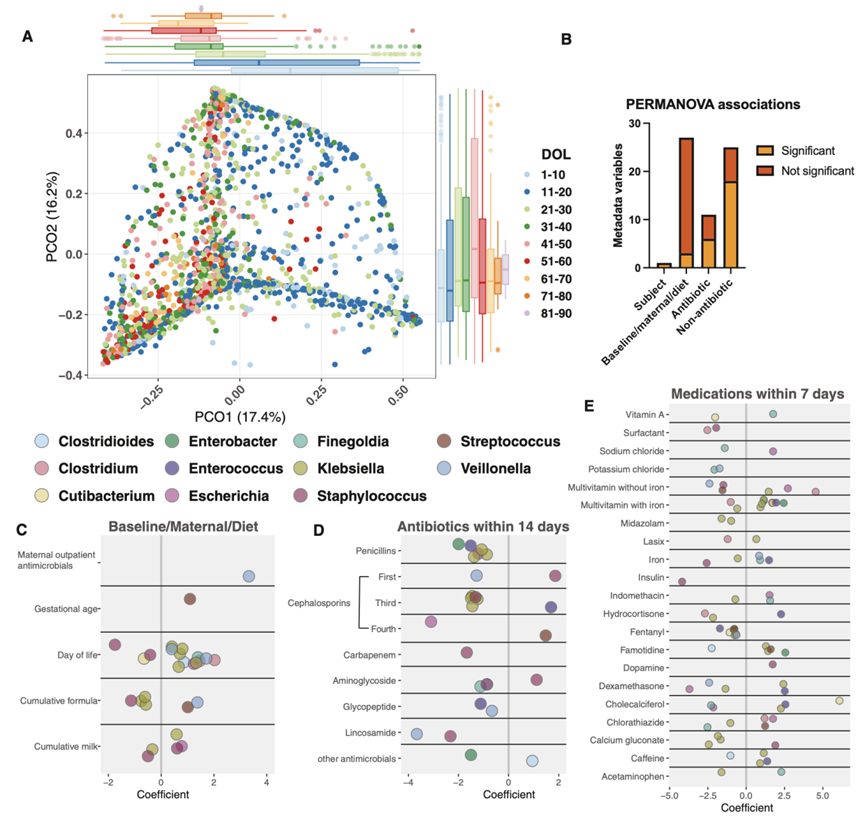
图3. NICU 暴露对早产儿肠道微生物群发育的影响。
(A) 188 名婴儿在 NICU 住院前 90 天内的 1888 个样本的 Bray-Curtis 差异性 PCoA。 (B) 通过重复测量 PERMANOVA 确定元数据变量类别的显著和不显著关联。(C-E)各属对应物种的变化系数。
04
早产肠道微生物组编码的功能发展轨迹
为了更好地了解肠道微生物组发育的动力学,他们建立了早产肠道微生物群功能组装的详细轨迹。为此,他们将宏基因组微生物组功能谱映射到来自相同粪便的 1381 个宏转录组中,以捕获编码代谢潜力的转录。通常,分类潜力和表达与早产肠道微生物组的分类组成相对应,通路丰富度和转录在出生后发育过程中增加。然而,他们发现不同功能组的转录存在相当大的差异。核心功能组的表达,包括几乎所有微生物组中都存在的糖酵解和核苷/核苷酸生物合成,在出生后发育过程中的变化小于编码氨基酸生物合成、碳水化合物降解或脂肪酸和脂质生物合成的基因表达(图 4 A)。至关重要的是,微生物群对氨基酸和脂肪酸的生物合成对宿主的免疫调节和发育至关重要,强调了这些功能组在出生后成熟过程中的转录轨迹可能直接影响婴儿的健康。在出生后发育过程中变化的通路表达中,肠杆菌科占主导地位,而大肠杆菌属、克雷伯菌属和Enterobacter spp.的基因组影响这些通路的转录(图4B-C)。相反,在刚出生后几周内,韦荣球菌和梭菌属在内的关键共生菌的转录贡献不太明显。
由于有证据表明 NICU 暴露组塑造了早产肠道微生物群的分类组成(图 3),他们假设环境也会影响编码功能轨迹的基因的转录。因此,他们开展了如上所述的 GLMM研究,并全面描述了 NICU 暴露组对代谢通路的影响。研究发现饮食因素、抗生素及非抗生素类药物会显著影响关键代谢单元(包括次级代谢产物、氨基酸、维生素和脂肪酸生物合成途径)的活性(图 4D)。虽然抗生素和大多数非抗生素药物与代谢潜力表达降低密切相关,但氯噻嗪和地塞米松与多种功能通路表达增加有关。重要的是,这种微生物组转录重构现象往往同时波及多个功能系统(图 4D),突显了 NICU 暴露组对早产肠道转录组的巨大的影响。
接下来,他们使用 GLMM 分析了分类单元对 NICU 暴露对 DNA 和 RNA 含量的响应之间的一致性。抗生素对转录的负面影响主要是由于菌群的减少而非调控引起的,但通路通常会因非抗生素药物和饮食暴露而下调(图 4 E),这表明肠道微生物组对早期生命暴露的反应是积极的转录调控。

图4. 肠道微生物组的转录轨迹分析。
(A) NICU 住院期间转录活性变化的特征。(B) 与出生后 DOL 相比,表达稳定或发生变化的通路中的物种富集。(C) 物种与功能类别的关联在出生后 DOL 中具有显著的表达变化。(D) NICU 暴露与通路表达变化显著相关。(E) 与选定暴露显著相关的通路表达变化热图,表明 DNA 丰度同时发生变化。
05
多组学分析支持变异微生物种群是 NEC 亚群的风险因素
早产肠道微生物组的异常发育进程在 NEC 的发展中起着根本性的作用。他们在 48 名婴儿中生成了肠道微生物组图谱。使用 1:2 匹配的病例对照研究设计进行比较(图 1 A),其中对照组根据 NICU 位置(圣路易斯、路易斯维尔和俄克拉荷马城)、GA(1 周内)和出生体重(100 克以内)与病例匹配。使用重复测量 PERMANOVA 和整个 NEC 队列标本,发病前 NEC 微生物组或宏转录组的分类组成及其功能谱均未因 NEC 状态而发生显著变化(图 5 A)。他们还发现在 NEC 发病之前任何分类群的细菌复制率没有显著差异(图 5 B)。同样,他们发现病例和对照的社区水平毒力因子库没有差异。此外,在 NEC 病例组中,微生物群转移既没有更频繁,也没有发生在相对于疾病发作的不同时间(图 5 C)。
为了分析多层次关联可能解释 NEC 个体化风险的可能性,他们使用三种不同的统计建模方法(逻辑回归、弹性网络和随机森林)结合了所有多组学数据(宏基因组、宏转录组、细菌复制率以及MAG 相关毒力谱)。至关重要的是,他们通过阻止个体模型交叉验证并在交叉验证过程中嵌套三次特征选择迭代(25、50 和 100 个特征)来考虑重复测量,分别对每个训练集执行特征选择。此外,他们使用所有 NEC 前数据、发病前 7 或 3 天或临床诊断当天收集的数据子集来训练模型。模型性能在算法之间或最终模型中包含的特征数量上没有差异(图 5 D)。他们观察到模型性能在诊断当天达到峰值,这表明在进行临床诊断时,病例和对照之间的微生物组特征更容易区分。
虽然不能可靠地预测所有婴儿的 NEC,但他们确实观察到,通过检测 NEC 发生的出生年龄,微生物群多样性存在显著差异(图 5 E)。具体而言,病例和对照组的微生物群多样性在出生后 40 天后分离,此时 NEC 病例的肠道微生物群与匹配对照组的肠道微生物群相比,多样性没有增加。12 名婴儿(占所有病例的 25%)在出生后 40 天后患上了 NEC,以下称为“晚发型”NEC。与出生后 40 天或以下发病的婴儿(“早发”)相比,晚发病例的出生胎龄明显较短,出生体重也较低。这与报告结果一致,即出生胎龄较短的婴儿与胎龄较长的婴儿相比,患 NEC 的时间更晚。
虽然在分别分析早发病例和对照时没有微生物组特征与 NEC 状态显著相关,但几种分类单元、通路和转录特征将晚发病例的微生物组与对照区分开来。对于晚发性 NEC,Negativicutes,特别是Veillonella parvula和 Clostridia 与对照状态显著相关,而Klebsiella 的丰度在事件发生前显著增加(图 5F-G)。当将 Dirichlet 多项式模型应用于病例和对照的分类肠道微生物群组成时,他们同样发现早发病例和对照无法区分,而晚发病例和对照的发育肠型轨迹有所不同。具体而言,与对照组相比,晚发性 NEC 病例中肠型 2-5 的比例过高。相反,所有晚发型 NEC 病例的粪便中均没有与肠型 6-8 一致的肠道微生物组组成。此外,与对照婴儿相比,晚发型 NEC 之前肠道微生物组的功能成熟度发生了显著改变(图 5 H)。这些特征在宏基因组水平上很明显,并且在细菌转录本分析中它们更加明显。因此,几种代谢组数据中,包括辅因子/辅基/电子载体/维生素生物合成、三羧酸 (TCA) 循环、次级代谢物降解和氨基酸降解,都表现出改变(与对照相比),而病例和对照之间在几种通路上没有潜在的宏基因组差异(图 5 H)。总的来说,这些数据表明,虽然微生物组可能不是所有早产婴儿个体化风险预测的关键,但它可能预测出生后 40 天后的疾病发作,并且宏转录分析比单独的宏基因组分析提供更多的鉴别信息。

图5. 肠道微生物组特征并不能预测所有早产儿 NEC 的发病情况,但可以区分晚发病例和对照。
(A) 肠道微生物组分类和功能组成的主坐标分析。(B) 在 NEC 发病前一周采集的样本中的细菌复制率。(C) 粪便样本中疾病发作前的微生物群转变事件的分布。(D) 受试者操作特征曲线分析。(E) Shannon 多样性。(F) 分类单元与晚发型 NEC 的病例或对照状态显著相关。(G) V. parvula和Klebsiella的丰度。(H) 与晚发型 NEC 相关的功能性通路表达和丰度。
+ + + + + + + + + + +
结 论
本项研究利用宏基因组学和宏转录组学方法,描述了三个新生儿重症监护病房住院的 236 名早产儿在出生后 3 个月内的肠道菌群动态。菌株追踪、分类学和功能分析以及全面的临床元数据表明,肠杆菌科、肠球菌和葡萄球菌主要利用可用的生态位来填充肠道菌群。艰难梭菌谱系在单个中心的个体之间持续存在,表皮葡萄球菌谱系在中心内和中心之间持续存在。总的来说,抗生素和非抗生素药物对肠道微生物组组成的影响大于母体或基线变量。最后,本项研究发现在出生后 40 天患上坏死性小肠结肠炎的新生儿体内存在持续的低多样性肠道微生物群。
+ + + + +



