

 English
English文献解读|Sci Adv(12.5):多组学整合揭示乳腺癌新辅助治疗反应的亚型特异性预测因子
✦ +
+
论文ID
原名:Multiomic integration reveals subtype-specific predictors of neoadjuvant treatment response in breast cancer
译名:多组学整合揭示乳腺癌新辅助治疗反应的亚型特异性预测因子
期刊:Science Advances
影响因子:12.5
发表时间:2025.07.04
DOI号:10.1126/sciadv.adu1521
背 景
乳腺癌是女性最常见的肿瘤类型,2020 年导致 685,000 人死亡。中国乳腺癌的年龄标准化发病率估计为每 100000 名女性 17.0 至 34.3 例。新辅助化疗 (NACT) 是一种广泛使用的乳腺癌术前治疗方法,旨在缩小肿瘤并使局部晚期或无法手术的乳腺癌可手术,避免腋窝淋巴结清扫并允许保乳手术。另一个重要优势在于有可能获得更多的预后和预测数据。鉴于残留癌症负担 (RCB) 与复发风险相关,可根据治疗反应定制治疗方案。例如,KATHERINE 试验表明,接受 HER2 靶向治疗后仍有残留病变 (RD) 的人表皮生长因子受体 2 (HER2) 阳性乳腺癌患者可从曲妥珠单抗美坦辛 (T-DM1) 治疗中获益,从而改善无复发生存期。虽然达到病理完全缓解 (pCR) 与更好的无事件生存期相关,但 NACT 可能会延迟手术并增加疾病进展风险。早期 pCR 预测可以降低这些风险并提高生存率。然而,NACT 反应背后的分子机制仍不清楚,准确的 pCR 预测仍然具有挑战性。
实验设计
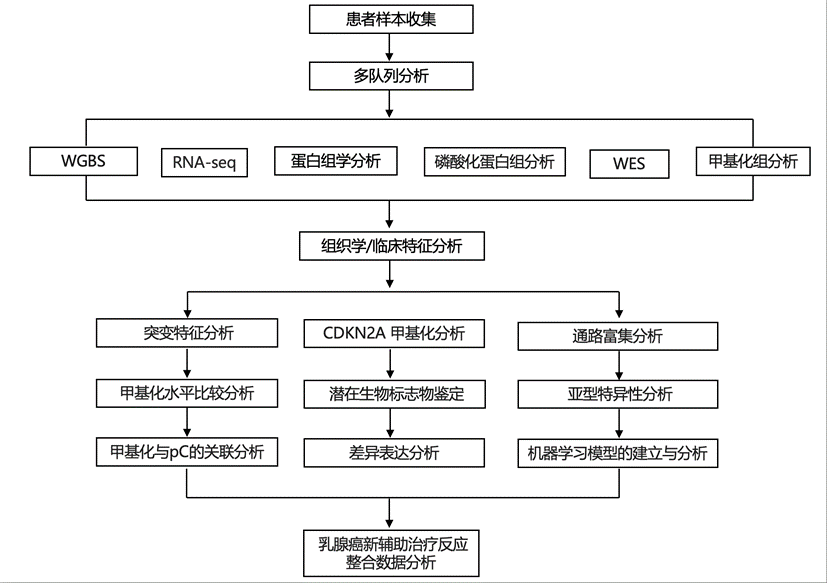
结 果
01

患者入组和治疗结果评估
为了研究乳腺癌治疗结果,研究团队从广东省人民医院 (GDPH) 招募了 149 名新诊断的患者(年龄范围:23 至 73 岁)(图 1A-B)。所有患者均接受了 NACT,然后进行了手术干预。使用免疫组织化学染色在诊断时评估 HER2 和雌激素受体 (ER) 状态,并将入组患者分为三个亚型:ER+ HER2 +(n = 55)、ER− HER2 +(n = 42)和 ER − HER2 −(n = 52)。除一例外,所有 HER2 +患者 均接受了抗 HER2 治疗。如图所示。 S1A,化疗方案包括多西他赛联合卡铂(TCb;n = 74)、表柔比星联合环磷酰胺联合多西他赛(EC-T;n = 61)以及多西他赛联合吡咯替尼(TY;n = 7)。另有7例患者接受了包括单用多西他赛(T)在内的方案。
将确定为接受 HER2 靶向治疗后仍有残留病变 (RD)的患者进一步分为 RCB 类,包括 RCB I、RCB II 和 RCB III。在 149 例患者中,81 例患者达到 pCR(ER+ HER2 + 26 例,ER− HER2 + 32 例,ER− HER2− 23 例),而 68 例患者为 RD(ER + HER2+ 29 例,ER− HER2+ 10 例,ER− HER2 − 29 例)。pCR 与 ER− HER2 −型总生存期改善以及 ER− HER2 −和 ER− HER2 +亚型良好无病生存期相关。接下来,他们使用多变量和单变量逻辑回归分析评估了临床特征与 pCR 之间的关联,发现与多种临床特征存在显著相关性(图 1C)。基于 HER2/ER 的亚型具有判别性 pCR 率:ER+ HER2 +为 47.3% ,ER− HER2 +为 76%,ER− HER2 −为 44% (图 1D)。随着肿瘤大小的增加,pCR 率从 T1 和 T2 中的 61% 下降到 T3 和 T4 肿瘤中的 17%(图1E)。此外,淋巴结转移的存在与 NACT 耐药相关,导致 pCR 的可能性较低,比值比 (OR) 为 0.39(图 1E)。此外,较高的肿瘤增殖标志物 Ki67 值与 ER − HER2−亚型的 pCR 率增加相关(图 1F)。
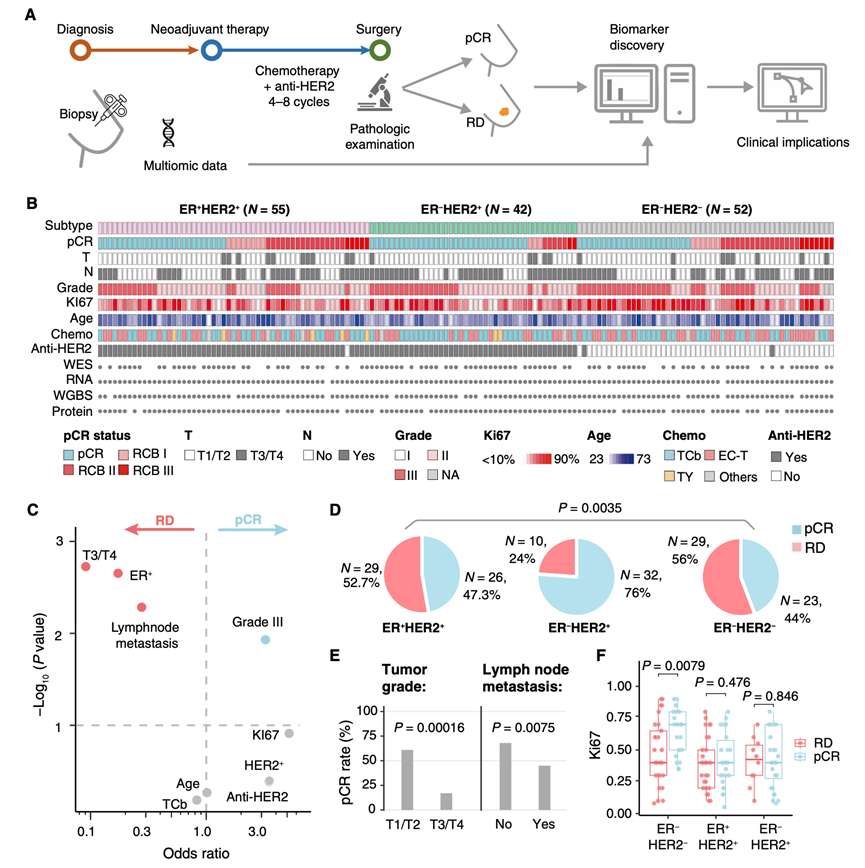
图1. 与 NACT 反应相关的队列和临床特征的总结。
(A) 本研究设计。(B) 本研究中使用的四种组学数据类型的临床信息和可用性摘要。(C) 通过逻辑回归进行多变量分析的结果,突出显示与 NACT 结果具有显著独立关联的临床特征。(D-E) pCR 与诊断时的亚型、临床肿瘤等级和淋巴结转移状态之间的关联。(F) 不同亚型间pCR与Ki67指数的关联。
02
乳腺癌样本的多组学分析
他们从治疗前肿瘤活检样本中收集了多组学数据(图 1B)。具体而言,对 104 对肿瘤和血液样本进行了全外显子组测序 (WES),以鉴定癌症基因组中的体细胞单核苷酸变异 (SNV)、小片段插入/缺失 (INDEL) 和体细胞拷贝数变异 (CNV)。此外,还分别对 145 份、140 份和 137 份肿瘤样本进行了全基因组亚硫酸氢盐测序 (WGBS)、转录组分析(RNA-seq) 和液相色谱-质谱分析,以分析甲基化组、转录组和蛋白质组/磷酸化组数据。在所有入组患者中,89 例拥有上述所有组学平台的数据。
使用GATK Mutect2对所有样本进行体细胞SNV和小INDEL检出,编码区非同义突变负荷中位数为74个(范围从3到1078个)。这些突变包括先前报道的乳腺癌驱动基因,例如TP53、PIK3CA、KMT2D、KMT2A和KMT2C、GATA3、MED13和CSMD1(图2A)。这些驱动基因的中位变异等位基因频率为 0.166。突变特征分析表明,年龄相关特征(SBS1 和 SBS5)和 APOBEC(载脂蛋白 B mRNA 编辑酶,催化多肽样)特征(SBS3 和 SBS13)占主导地位,这表明这些过程驱动了大部分观察到的诱变(图 2B)。正如预期的那样,SBS1 和 SBS5 与患者诊断时的年龄呈正相关。值得注意的是,SBS3 特征在 ER− HER2−中富集,但在 SBS13 中没有富集。随后,应用癌症重要靶点基因组识别2 (GISTIC2) 在该队列中发现了复发性局灶性和臂水平拷贝数变异。与先前结果一致, ERBB2、MYC、AIM1和CCND1的扩增在乳腺癌队列中大量存在(图2B)。
为了探究体细胞突变是否有助于治疗反应,他们比较了 pCR 组和 RD 组中非静默体细胞突变的频率。在校正错误发现率 (FDR) 后,体细胞基因 SNV/INDEL 与 pCR 无显著相关性。对于 CNV,通过比较各亚型中 pCR 组和 RD 组的局部 CNV 分布,他们发现ER− HER2 −患者群体中 RD 组的6q27拷贝数显著较低(图 2C-E)。进一步纳入更多样本或许可以验证这一观察结果,并能够识别更多可预测治疗结果的基因变异。
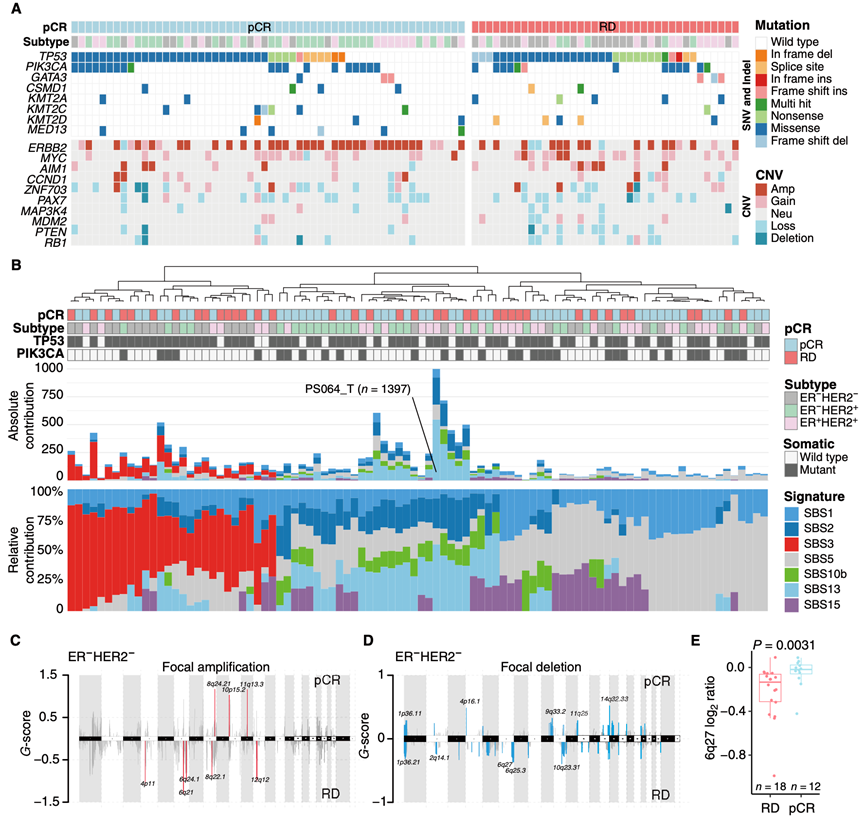
图2. 体细胞突变特征和景观。
(A) pCR 组和 RD 组体细胞突变情况。(B) 94 例患者的绝对和相对 SNV 标记分布。(C-D) ER− HER2 −亚型中 pCR 组和 RD 组之间的局部扩增和局部缺失比较。(E) ER− HER2−亚型中细胞带 6q27 的对数比率。
03
pCR 与 RD 乳腺癌样本的甲基化组比较
基因调控区甲基化在调控基因表达中起着至关重要的作用,并可能影响治疗反应。为了探究pCR与甲基化景观之间的关系,他们分析了145个肿瘤样本的WGBS数据,重点研究了预先定义的基因调控区及其与NACT结果的关联。初步分析显示,当亚型未分层时,pCR组和RD组之间的甲基化水平无显著差异。然而,与先前的研究一致,他们观察到HER2+亚型启动子和增强子区域的甲基化水平显著高于ER-HER2-亚型(图3A)。在比较各亚型中pCR组和RD组的甲基化水平时,ER+HER2+亚型中远端增强子样标记区域和CCCTC结合因子(CTCF)特异性区域的甲基化水平升高(图3A)。这些发现提示可能存在与治疗反应相关的亚型特异性甲基化模式,值得进一步研究(图3A)。
为了揭示 pCR 和 RD 之间更具体的甲基化差异,他们将 WGBS CpG 甲基化数据分割成甲基化区域以进行差异甲基化分析(图 3B)。为了解决 CpG 甲基化数据的高维性并减少假阳性,他们开发了一种基于网络的特征选择方法,该方法整合了来自生物途径、基因-疾病关联、蛋白质-蛋白质相互作用和基因调控的先验知识,以构建乳腺癌特异性生物网络,称为 BRCANET(图 3B)。他们进一步将甲基化区域与相应的基因表达相关联,以识别甲基化调控区域,优先考虑那些具有显著的甲基化表达负相关性和 pCR 和 RD 之间差异甲基化的区域(图 3B)。使用此框架,突出显示了 BRCANET 背景下的甲基化区域候选者。
他们将全基因组CpG甲基化数据划分为多个区域,并根据RNA甲基化关联和按NACT结果分层的甲基化差异,选择每个基因中排名靠前的差异甲基化区域(DMR)作为其代表[差异甲基化基因(DMG)]。对所有亚型进行综合分析时,发现FGF1和SNAI1基因的高甲基化与pCR组相关,而MAP4K1和CDKN2A基因的高甲基化则见于RD组。分别在 ER− HER2 −、ER − HER2 +和 ER + HER2 +亚型中,他们在 pCR 组中鉴定出 465、576 和 2242 个高甲基化基因,而在 RD 组中分别鉴定出 1381、708 和 208 个高甲基化基因。使用 BRCANET 框架,进一步将这些发现细化为 ER − HER2 −、ER − HER2 +和 ER + HER2 +亚型中 pCR 组中的 37、32 和 98 个高甲基化基因以及RD 组中的 69、15 和 11 个高甲基化基因(图 3C)。值得注意的是,参与细胞周期的CDKN2A和BIRC5在 ER− HER2 − RD 肿瘤中显著高甲基化。在 RD 肿瘤中,ER− HER2 +中的XRCC2和ER+ HER2 +中的MAP4K1和MFNG均发生高甲基化(图 3C)。相反,NFIA(核因子 IA)、BRCA1(BRCA1 DNA 修复相关,参与 DNA 修复)和SNAI1 (蜗牛家族转录抑制因子 1,参与上皮-间质转化)高甲基化与 ER− HER2 −肿瘤中的 pCR 相关; GADD45A(生长停滞和 DNA 损伤诱导 α)、NCOR2(核受体辅抑制因子 2)、SNAI1和KIT(受体酪氨酸激酶)在 ER−HER2 +肿瘤中高甲基化;AQP1(水通道蛋白 1)、PPARA、SPOP和JUP(编码一种常见的连接斑块蛋白)在ER+HER2+ 肿瘤中发生高甲基化(图 3C)。
CDKN2A是一种已知的抑癌基因,在各种癌症中经常通过启动子高甲基化等机制失活。为了进一步证实CDKN2A DMR 甲基化作为 ER− HER2 −患者 pCR 的预测因子,他们收集了一个接受 NACT 治疗的 75 例 ER − HER2 −患者的独立队列,根据发现队列中的甲基化谱设计了一个亚硫酸氢盐焦磷酸测序区域,使用 100 bp 的滑动窗口。这种方法使他们能够识别出与 ER− HER2−肿瘤 pCR 最密切相关的优化 CpG 区域,该区域位于CDKN2A的一个转录起始位点 (TSS) 附近(图 3D)。然后对从另外收集的 75 例病例中提取的福尔马林固定石蜡包埋 (FFPE) 样本进行测序。基于WGBS数据与焦磷酸测序数据的一致性以及测序质量进行质量控制后,他们选取一个CpG位点(9号染色体:21996194)进行下游分析。虽然样本量较小且样本为FFPE,但在验证队列中观察到高甲基化与RD之间存在显著关联(图3E)。
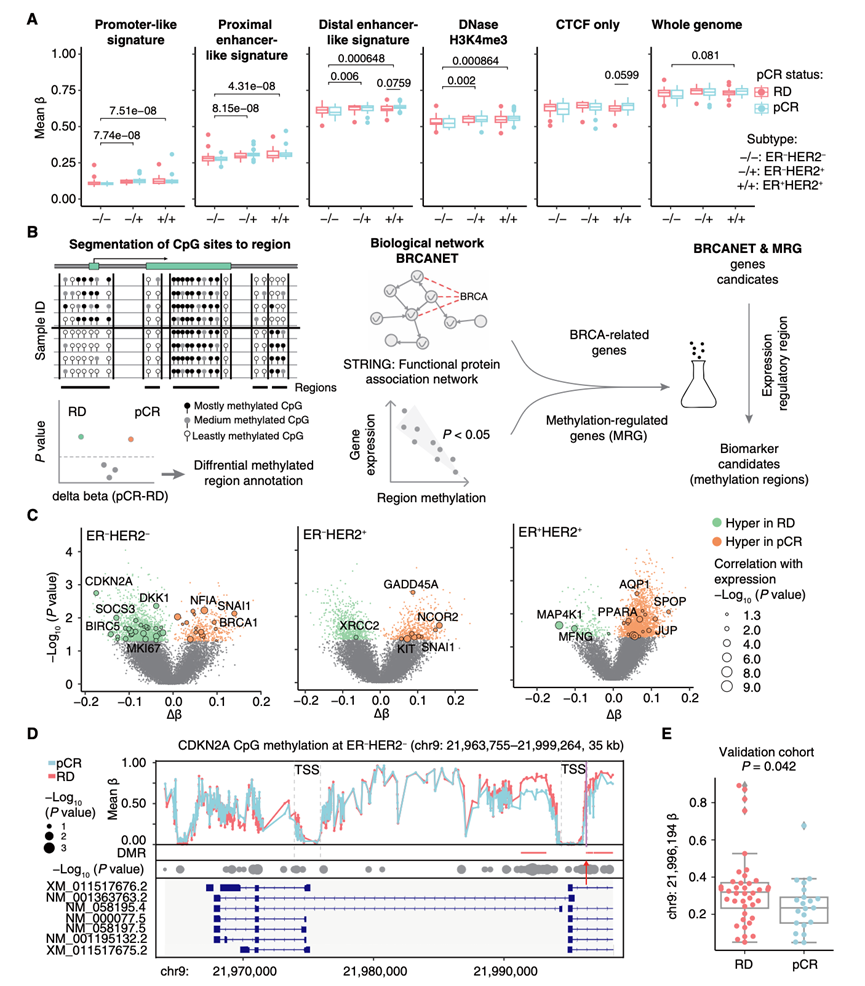
图3. 甲基化特征作为 pCR 的预测因子。
(A) 各亚型中 pCR 和 RD 顺式调控元件的平均甲基化水平。(B) 识别甲基化生物标志物的框架。(C) 各亚型中 pCR 与 RD 的 DMG。(D) ER− HER2−中CDKN2A的 CpG 水平甲基化。(E) 通过焦磷酸测序得到的 chr9: 21996194 处的甲基化水平分布。
04
亚型相关的化学敏感性转录组预测因子
他们利用140例患者的RNA-seq数据,进一步探究了pCR与基因或通路表达之间的关联。首先,通过t分布随机邻域嵌入(t-SNE)检验基因表达的分布,发现属于同一亚型的样本呈现出聚集在一起的趋势(图4A)。对t-SNE1和t-SNE2组分分布的检验,揭示了不同亚型之间存在显著差异。按pCR和RD分层后,仅t-SNE2在ER− HER2 +亚型中表现出显著差异。接下来,为了创建与临床表型相关的基于表达的聚类,他们首先进行了三向方差分析 (ANOVA),将基因表达与 HER2 状态、ER 状态和 pCR 状态相关联,然后使用 ANOVA 识别的基因实施层次聚类(图 4B)。
接下来,他们使用 DESeq2进行差异表达分析,在 pCR 组和 RD 组中分别鉴定出 1481 个和 1431 个过表达基因。BRCANET 分析突出显示了 84 个 pCR 相关和 45 个 RD 相关过表达基因,其中包括ERBB2,它在 pCR 中过表达。基因集富集分析 (GSEA)显示,pCR 相关基因在增殖相关通路(例如 MTORC1_SIGNALING 和 E2F_TARGETS)和免疫相关通路(例如 ALLOGRAFT_REJECTION 和 INFLAMMATORY_RESPONSE)中富集。亚型特异性分析在 ER− HER2−、ER− HER2+ 和 ER + HER2+ 肿瘤中分别鉴定出 pCR 组中 911、782 和 584 个过表达基因,在 RD 组中分别鉴定出 905、1056 和 564 个过表达基因(图 4C)。Wilcoxon 秩和检验和 BRCANET 在 ER− HER2−、ER − HER2 +和 ER + HER2+肿瘤中分别突出显示出59、38和19个pCR相关过表达基因以及 34、39 和 20 个 RD 相关过表达基因(图 4C)。
在 ER− HER2 −肿瘤中,pCR 与KLK3和AR表达降低相关,但与CDK1和MKI67等增殖相关基因表达升高相关。在 ER− HER2 +肿瘤中,RD 患者的GFRA3和FOXO3表达升高,而在 ER+ HER2 +肿瘤中,RD 患者的ABCC3表达升高,ABCC3 是一种与多药耐药相关的 ABC 转运蛋白基因(图 4C)。通路富集分析显示,在 pCR 组的不同亚型中,细胞周期通路(例如,E2F_TARGETS、G2M_CHECKPOINT、MTORC1_SIGNALING)和免疫相关通路(例如,MYC_TARGETS_V1 和 INTERFERON_ALPHA_RESPONSE 和 INTERFERON_GAMMA_RESPONSE)均富集,且其显著性水平因亚型而异(图 4D)。
总体而言,转录组分析显示,在达到pCR的ER - HER2-肿瘤中,增殖相关基因和通路表达升高。pCR组和RD组之间的差异表达基因和通路在不同分子亚型之间有所不同,提示存在潜在的亚型特异性治疗耐药或敏感性机制。
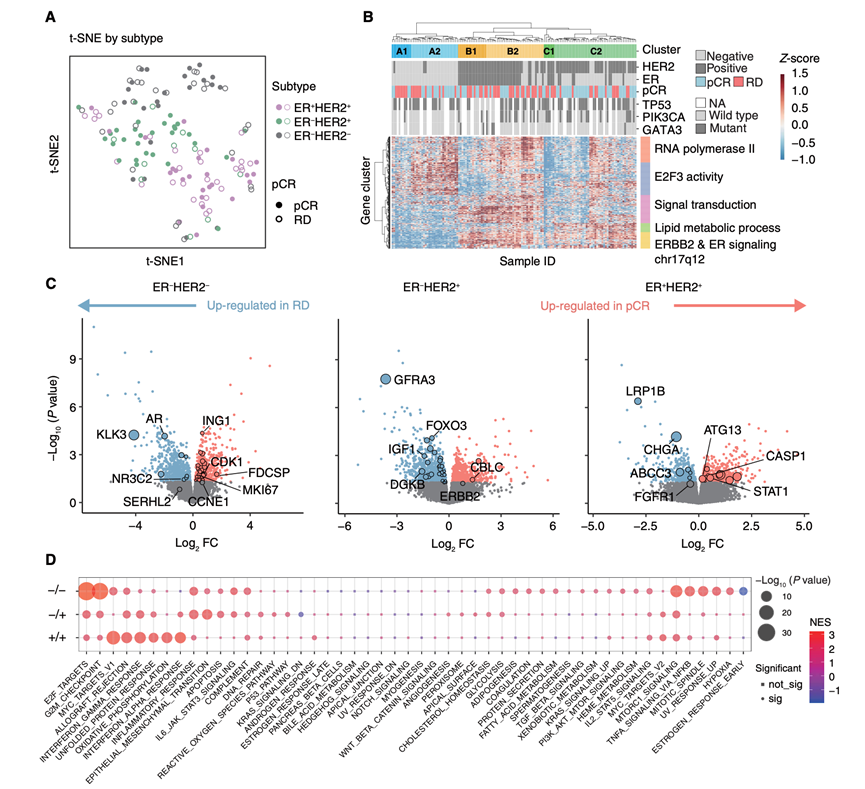
图4. 与 NACT 结果相关的转录组特征。
(A) 基因表达的 t-SNE 分析。(B) 基于方差分析 (ANOVA) 的基因表达聚类。(C) 各亚型中 pCR 和 RD 之间的差异表达基因。(D) 对各亚型的标志性通路进行基因集富集分析 (GSEA)。
05
蛋白质组学分析发现TOP2A是三阴性肿瘤的预测因子
为了揭示pCR患者和RD患者之间的转录后差异,他们分析了137例个体的蛋白质和磷酸化蛋白表达数据,比较了5473个蛋白质,鉴定出pCR组中152个过表达,RD组中156个过表达。BRCANET突出显示了13个pCR相关蛋白[例如,MCM7(微染色体维持复合物组分7)、ERBB2(erb-B2受体酪氨酸激酶2)和S100A8(S100钙结合蛋白A8)]和8个RD相关蛋白[例如,VCAN(多功能蛋白聚糖)、PTPRG(蛋白酪氨酸磷酸酶受体G型)和EGFR(表皮生长因子受体)]。在 1737 种磷酸化蛋白中,42 种在 pCR 组中过表达[例如,RPS6KB1(核糖体蛋白 S6 激酶 B1)、FBLN2(原纤维蛋白原 2)、MCM2(微染色体维持复合物成分 2)和 ABCC1(ATP 结合盒亚家族 C 成员 1)],而 45 种在 RD 组中过表达[例如,HIC1(HIC ZBTB 转录抑制因子 1)、FBXO4(F-box 唯一蛋白 4)和 CREBBP(CREB 结合蛋白)]。亚型特异性分析揭示了与 pCR 相关的差异表达蛋白。在 ER− HER2−肿瘤中,BRCANET 突出显示了 27 种 pCR 相关蛋白[例如 MCM7、CDK1(细胞周期蛋白依赖性激酶 1)、PCNA(增殖细胞核抗原)、MCM2 和 TOP2A(DNA 拓扑异构酶 II α)]和 20 种 RD 相关蛋白(例如 HERC2,一种 DNA 损伤反应调节剂)(图 5A)。在 ER− HER2 +肿瘤中,CTR9 和 HIF1AN 在 pCR 组中过表达,而乳腺癌细胞迁移所必需的 STAT5B 和参与 DNA 错配修复的 MSH6 在 RD 患者中升高。具体而言,MSH6 蛋白表现出与更高的 RCB 状态相对应的表达增加趋势(图 5A)。在 ER+ HER2 +肿瘤中,BRCANET 突出显示了 8 种 pCR 相关蛋白和 5 种 RD 相关蛋白(图 5A)。
对于差异表达磷酸化蛋白 (DEPP),ER−HER2−肿瘤在 pCR 组中表现出细胞周期相关蛋白 (例如 TOP2A、MKI67、MCM2 和 RPS6KB1)的过度表达,而 RD 患者的 HSBP1、HIC1 (与染色体稳定性相关)和 MTOR(参与应激反应)表达较高(图 5B)。在 ER− HER2 +和 ER + HER2 +肿瘤中,仅鉴定出少数 DEPP,例如 ER − HER2 +中的 FBXO4 和 ADAM17 以及 ER + HER2 + RD患者中的 SMARCA2 和 CTNND1(图 5B)。
使用蛋白质和磷酸化蛋白丰度进行标志通路分析显示,ER− HER2− pCR 患者细胞周期相关通路(例如 E2F_TARGETS 和 G2M_CHECKPOINT)表达较高,而 RD 患者脂肪生成和肌肉生成通路表达增高,蛋白质和磷酸化蛋白数据均支持这一观点(图 5C)。脂肪生成蛋白通路表达增高在独立的 ER− HER2 −队列中得到了独立验证。
TOP2A是一种 DNA 拓扑异构酶,在 ER− HER2 − pCR患者的蛋白质和磷酸化蛋白水平上均显著上调,且在 RNA、蛋白质和磷酸化蛋白水平上呈现相关的表达模式。其过表达与增强的蒽环类药物治疗反应有关。鉴于多组学支持的TOP2A差异,他们研究了多组学整合是否有助于识别 pCR 患者和 RD 患者之间的其他趋同差异。他们对 pCR 和 RD 条件下不同乳腺癌亚型中显著差异表达的基因进行了多组学整合和 Hallmark 通路富集分析(图 5D)。不同亚型之间的通路富集模式差异很大。在 ER− HER2 −亚型中,多组学和单组学分析均鉴定出显著富集的通路,主要与细胞周期调控有关(例如 E2F_TARGETS、G2M_CHECKPOINT 和 MITOTIC_SPINDLE)。例如,他们可以观察到 E2F_TARGETS 通路中的基因在各种单组学数据集中表现出显著且方向一致的变化(图 S13D)。相反,ER− HER2 +和 ER + HER2+肿瘤显示出更分散的通路富集。通过多组学整合,他们观察到了潜在的生物学相关信号,例如 ER− HER2 +肿瘤中的 XENOBIOTIC_METABOLISM 通路,这在单组学分析数据中并不显著(图 5D)。
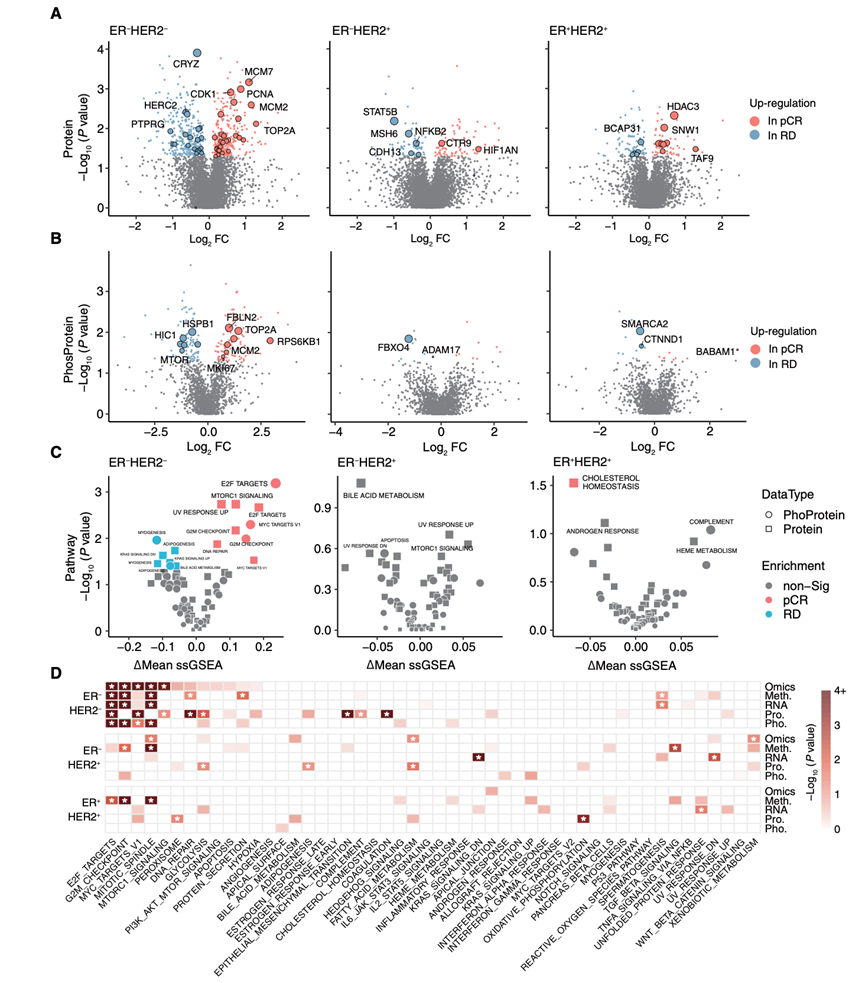
图5. 与 NACT 结果相关的蛋白质组学特征。
(A-B) 每个亚型中 pCR 和 RD 之间的差异表达蛋白质和磷酸化蛋白。(C) pCR 和 RD 之间的蛋白质和磷酸化蛋白通路差异。(D) 通路富集分析。
06
预测患者结果的综合模型
他们使用 LightGBM 算法开发了模型,并通过 30 次重复的五重交叉验证评估了 MOPCR 相对于单组学模型的性能(图 6A)。MOPCR 在所有亚型中始终优于单组学模型(图 6B)。在 ER− HER2 −亚型中,MOPCR 的平均准确率为 0.82,AUC 为 0.98,精确召回曲线下面积为 0.97,明显超过单组学模型。在 ER− HER2 +和 ER + HER2 +亚型中也观察到了类似的改善(图 6B)。
为了确保可解释性,他们计算了 SHapley Additive exPlanations (SHAP) 值来评估 MOPCR 中特征的重要性(图6C)。ER− HER2 −模型的关键特征包括 MTORC1_SIGNALING、E2F_TARGETS 和 UV_RESPONSE_UP 蛋白通路、KI67 指数和CDKN2A甲基化。在 ER− HER2 +模型中,KIT甲基化状态是最关键的特征,其次是肿瘤等级、肿瘤大小和 RNA 水平通路,包括 EPITHELIAL_MESENCHYMAL_TRANSITION 和 INFLAMMATORY_RESPONSE。对于 ER+ HER2 +模型,MAP4K1甲基化水平是最关键的特征,其次是 RNA 水平的 MYC_TARGETS_V1、磷酸化蛋白水平的 UNFOLDED_PROTEIN_RESPONSE 和蛋白质水平的 HEME_METABOLISM 通路。值得注意的是,甲基化特征在ER− HER2 +和ER+HER2+亚型中始终位居最显著的预测因子之列(图6C),凸显了甲基化分析在治疗结果预测中的重要性。需要在更大、更多样化的队列中进一步验证,以证实这些发现,并探索甲基化标记与治疗结果之间的因果关系。
MOPCR 证明,在焦磷酸测序队列中,整合CDKN2A甲基化和 Ki67 指数的效果优于仅使用单一特征的效果,强调了多组学整合的价值。为了进一步评估稳健性和普遍性,他们将 MOPCR 应用于公开可用的数据集。对于缺乏甲基化数据的数据集,他们使用基于 RNA 数据的最小绝对收缩和选择算子 (LASSO) 模型来估算甲基化水平(图 6A)。在 TransNEO 数据集中,MOPCR 使用估算甲基化比使用 RNA 替代甲基化表现出更好或相当的性能。在三个独立的 RNA-seq数据集中,ER− HER2 −亚型的 AUC 值范围为 0.589 至 0.768。ER− HER2+和 ER+ HER2 +亚型也有类似的表现(图 6D)。 BrightNess 研究中,纳入了三种治疗方案——(A)紫杉醇 + 卡铂 + 维利帕尼;(B)紫杉醇 + 卡铂 + 安慰剂;以及(C)紫杉醇 + 安慰剂 + 安慰剂——MOPCR 在 A、B 和 C 组中分别实现了 0.61、0.60 和 0.55 的 AUC 值。A 组和 B 组(包括卡铂(一种 DNA 复制靶向药物)和维利帕尼(一种 DNA 修复靶向药物))的性能改善表明,MOPCR ER− HER2 −模型可能更有助于评估紫杉醇与其他细胞周期靶向药物相结合的治疗方案。
在所有接受 MOPCR 预测 pCR 概率的独立 RNA-seq 队列中,ER− HER2−亚型在 pCR 组中表现出的可能性明显高于 RD 组,虽然RD 组中存在 pCR 概率得分高而 pCR 组中 pCR 概率得分低的情况。未来使用更大的数据集整合更多高置信度生物标志物可能有助于提高预测性能。为了方便研究界使用 MOPCR 实现乳腺癌 NACT pCR 预测的长期愿景和前景(图 6E),他们开发了一个用于部署原型模型的网站,可通过http://www.wang-lab-hkust.com:8050. 进行访问。总之,虽然仍需要进一步研究,但本研究研究表明,使用机器学习技术结合多组学数据和临床信息可以对 pCR 做出有益的预测。
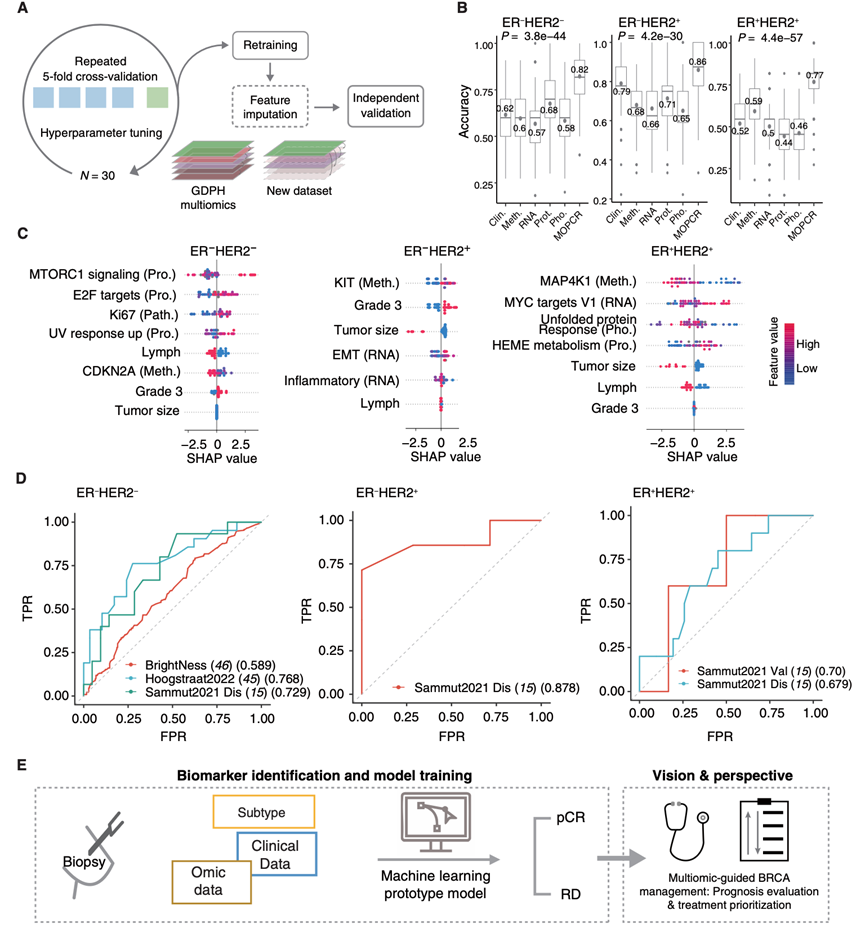
图6. 临床结果的机器学习模型预测分析。
(A) 用于训练和验证机器学习模型的框架。(B) 三种亚型中单组学模型和多组学 pCR 预测模型 (MOPCR) 的交叉验证性能(准确性)比较。(C) 优化的 MOPCR 模型中特征的 SHapley Additive exPlanations (SHAP) 值。(D) 用于在外部数据集中针对每个亚型进行独立验证的接收者操作特征曲线。(E) 本研究中机器学习预测因子的总结。
+ + + + + + + + + + +
结 论
本研究对 149 名中国乳腺癌患者的肿瘤样本进行了多组学分析,涵盖 ER− HER2+、ER+ HER2+和 ER− HER2−亚型,将结果分为病理pCR或RD,发现每种亚型都有与 pCR 相关的独特分子特征:ER−HER2−pCR 患者的细胞增殖增加,ER− HER2 − RD患者的CDKN2A甲基化增加,ER− HER2 + RD患者的KIT甲基化增加,以及ER+ HER2 + RD患者的MAP4K1高甲基化。这些发现随后在独立数据集中得到验证。通过整合临床和多组学数据,本研究开发了MOPCR,这是一种针对亚型的机器学习模型,其在预测治疗反应方面优于单组学方法。MOPCR展现了跨队列的潜在通用性,并对具有较高pCR概率的患者亚组进行了初步分层,为精准癌症管理提供了宝贵的见解。
+ + + + +



