

 English
English文献解读|Cell(42.5):功能性肝脏基因组学鉴定出促进癌症恶病质消瘦的肝细胞因子
✦ +
+
论文ID
原名:Integrated single-cell and spatial transcriptomics uncover distinct cellular subtypes involved in neural invasion in pancreatic cancer
译名:整合单细胞和空间转录组学揭示与胰腺癌神经侵袭有关的不同细胞亚型
期刊:Cancer Cell
影响因子:44.5
发表时间:2025. 07.17
DOI号:10.1016/j.ccell.2025.06.020
背 景
癌症能够引发患者全身代谢的重大变化。这种现象在癌症恶病质 (CCx) 的背景下尤为明显,这是一种多因素综合征,其特征是体重不由自主地大幅下降。根据肿瘤类型,50%–80% 的癌症患者患有 CCx。它由全身炎症、能量消耗增加、分解代谢增加和食欲不振等多种因素共同引起,这些因素均由肿瘤和宿主源性因素引起。CCx会导致进行性功能障碍、生活质量下降、化疗毒性增加以及死亡率增加。CCx至少占癌症相关死亡的 20%,目前,尚无美国食品药品监督管理局 (FDA) 批准的可以完全逆转 CCx 的治疗方法。
实验设计
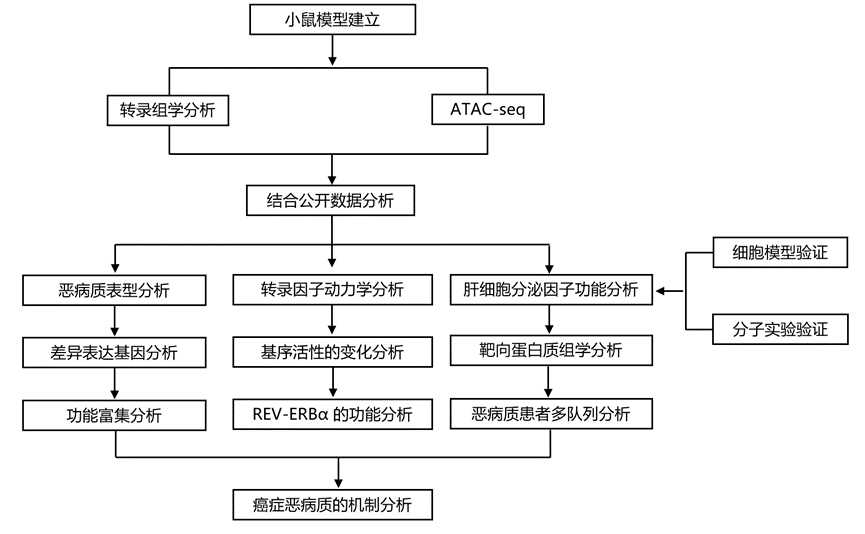
结 果
01

肝脏转录组分析可识别恶病质相关程序
为了研究CCx中肝细胞的特异性变化,研究团队使用“特定细胞类型标记细胞核分离”(INTACT)方法,从不同体重稳定型癌症小鼠模型和CCx小鼠模型的肝细胞中分离细胞核。INTACT方法允许进行Cre-lox驱动的细胞类型特异性标记,并随后从INTACT组织中对细胞核进行亲和力纯化。他们将诱导恶病质的癌细胞(C26 结肠癌和 8025 胰腺导管腺癌 [PDAC])或导致体重稳定型癌症的癌细胞(NC26 和 MC38 结肠癌细胞)植入同基因肝细胞完整型 (HEP-INTACT) 小鼠体内,该小鼠特异性地在肝细胞中表达核标签 (sun1 sfGFP Myc)。根据癌细胞系的不同,使用 BALB/c 或 C57BL/6 小鼠作为实验背景(图 1 A)。
正如预期的那样,与对照组相比,恶病质 C26 (C26_Cx) 和 PDAC 8025 肿瘤小鼠体重明显下降(图 1B),腓肠肌 (GC)、附睾白色脂肪组织 (eWAT) 和皮下 (sc) WAT 重量也减轻。他们还纳入了恶病质前期 C26 肿瘤小鼠 (C26_pre-Cx),与对照组相比,其体重没有明显变化(图1B),代表消瘦前状态。在这一组中,肿瘤重量低于恶病质 C26 肿瘤小鼠。相比之下,NC26 和 MC38 肿瘤没有引起体重或组织重量的显著变化(图1 B),证实了这些癌症模型的体重稳定性。
他们从这些小鼠中分离出细胞核,并对整个肝脏和 GFP+ 肝细胞核进行了转录组分析 (RNA-seq),并对肝细胞核进行了转座酶可及染色质测序 (ATAC-seq) 测定(图 1 A)。主成分分析 (PCA) 和相关性分析显示,体重稳定肿瘤组(NC26 和 MC38)与注射 PBS 的对照组相比,总体变化较小(图 1 C)。相比之下,两种恶病质模型(C26 和 8025)的表达和 ATAC-seq 谱均显示出显著且明显的改变,其中恶病质前组的表现介于对照组和完全恶病质组之间(图 1 C)。
正如预期的那样,与整个肝脏细胞核相比,肝细胞标记基因在 GFP+ 肝细胞核中富集,相反,非实质肝脏驻留细胞和炎症细胞类型的标记基因表达更高(图 1 S-T)。为了阐明肝脏对恶病质进展的转录反应,他们重点关注来自C26 模型(该模型包含一个恶病质前时间点)和相应的体重稳定 NC26 模型的数据,鉴定出 5534 个基因,这些基因在 GFP+ 肝细胞核或全肝细胞核中的一种或多种条件之间发生显著改变,并对这些基因进行 K 均值聚类(图 1D)。他们鉴定出 9 个基因聚类(GC1-GC9)。其中六个在纯化的肝细胞核(GC1–GC6)中显示信号增强,而其余三个(GC7-GC9)中的基因主要在全肝细胞核中表达。在体重稳定(MC38)和恶病质癌症(8025)两种互补模型中,这些基因聚类相对于 PBS 注射小鼠始终表现出相似的表达变化,包括在恶病质小鼠中诱导基因聚类 3(GC3)。
GO分析确定了在单个聚类中富集的功能类别(图 1 E)。恶病质进展期间在肝细胞中激活的基因 (GC3) 在氨基酸代谢/分解代谢过程和急性期反应通路中富集,而恶病质进展与参与蛋白质折叠 (GC4) 和脂肪酸代谢 (GC6) 的肝细胞基因的抑制有关。在也包含非实质细胞 (NPC) 细胞核的全肝细胞核中表达增加的基因在与免疫过程和细胞外基质组织相关的方面表现出富集 (GC8),而恶病质抑制的基因似乎参与了内皮细胞迁移 (GC9)。根据这些通路分析,他们发现特别是 GC3 和 GC8 包含许多编码分泌蛋白的基因。此外,GC3 含有编码众所周知的分泌性急性期反应蛋白的基因,例如血清淀粉样蛋白 A1 和 A2 (Saa1和Saa2)、α-1-酸性糖蛋白 1-3(Orm1、Orm2和Orm3)和纤维蛋白原(Fga、Fgb和Fgg)(图 1F)。他们使用 NicheNet方法对基因调控网络中配体与其假定靶基因之间的相互作用进行预测,以揭示肝细胞和全肝细胞核内 CCx 基因程序改变的潜在上游调节因子(图 1G)。在肝细胞中,该分析强调了促恶病质 IL-6、制瘤素 M(OSM)、IL-27、IL-22 和 IL-22b 是主要的上游调节因子。在整个肝细胞核中,除了大量白细胞介素外,他们还发现肿瘤坏死因子 α (TNF-α) 和干扰素 γ 是之前与 CCx 相关的调节基因表达变化的因子。综上所述,他们发现CCx肝细胞的转录反应与体重稳定的癌症不同,且涵盖了多种肿瘤模型。此外,肝细胞中编码分泌蛋白的基因显著上调,提示肝细胞可能通过某种途径影响恶病质的进展。
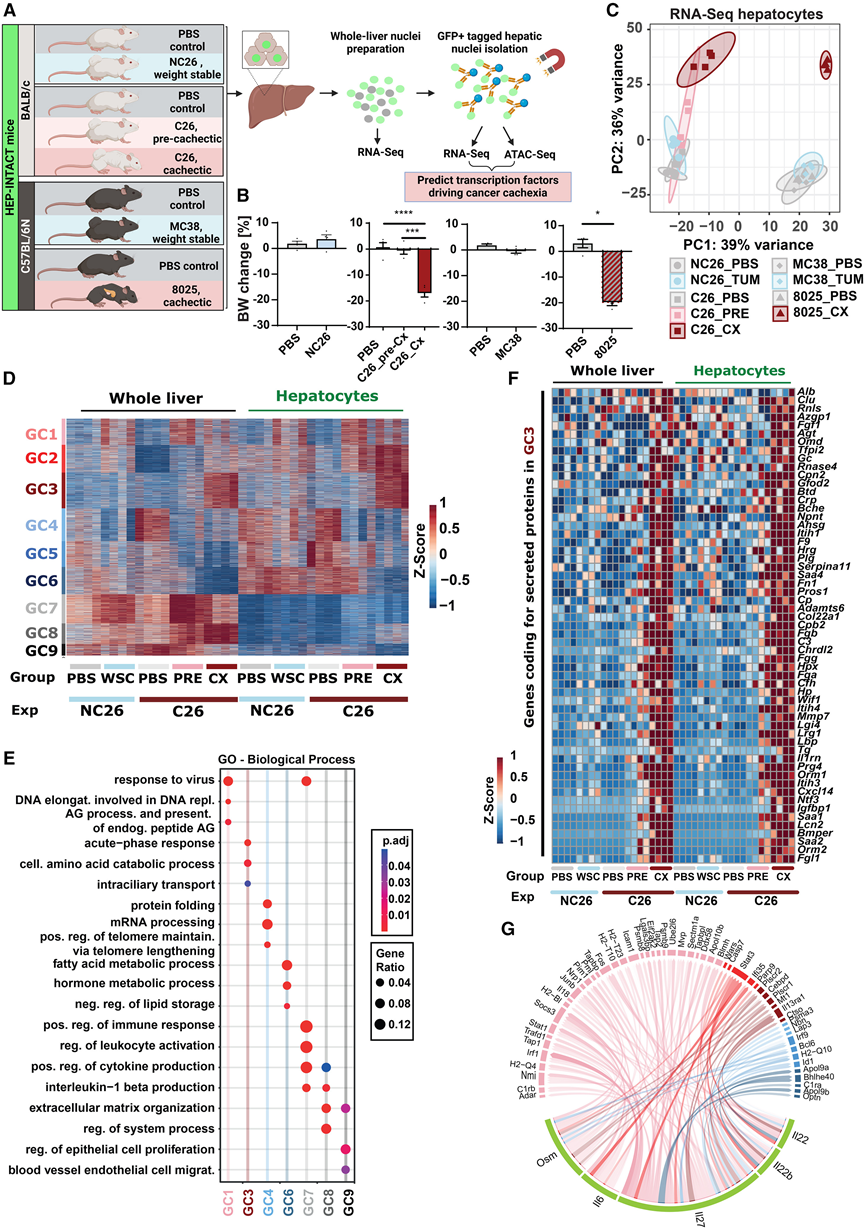
图1. 癌症恶病质中肝脏转录组的时间动态。
(A) 实验流程。(B) 体重变化。(C) 肝细胞RNA-seq数据的主成分分析。(D) 差异表达基因的行尺度表达进行 K 均值聚类。(E) 功能富集分析。(F) 热图显示基因聚类 3 内编码分泌蛋白的基因的行尺度表达。(G) NicheNet 分析恶病质期间肝细胞中的配体-靶标连接。
02
增强子和转录因子动力学分析表明 REV-ERBα 是肝细胞恶病质反应的调节因子
为了探究恶病质进展过程中肝细胞中观察到的基因表达变化背后的调控机制,他们接下来分析了从对照小鼠和荷瘤小鼠肝脏中分离的 GFP+ 肝细胞核的 ATAC-seq 数据。首先关注 C26 和 NC26 模型,鉴定出 13906 个增强子,它们在 GFP+ 肝细胞核中的一种或多种条件下染色质可及性发生显著变化,并对这些增强子进行 K 均值聚类,鉴定出四个具有不同时间特征的增强子聚类 (EC),体重稳定的癌症和恶病质的影响在 MC38 和 8025 模型中通常是保守的。这些 EC 在恶病质进展过程中具有相似时间激活模式的表达聚类基因附近富集 (图 2B),例如Orm2/3、Igfbp1、Fabp1和Ces1f基因座(图 2C)。在这些情况下,恶病质期间的晚期基因激活与附近增强子 (EC2) 处染色质可及性的晚期增加相关(图 2C),或恶病质期间的抑制与附近增强子(EC3 和 EC4)处染色质可及性的降低相关。
为了鉴定在恶病质发病机制中驱动肝细胞基因表达变化的转录因子,他们使用了“转录因子基序活性和基因表达变化整合分析”(IMAGE)机器学习方法来预测哪些转录因子基序导致了观察到的转录活性变化(调控基序活性)。他们最初关注 C26 和 NC26 模型,鉴定出98个在 GFP+ 肝细胞核中一种或多种条件下活性发生显著变化的基序(图 2D)。这些基序分为四个基序聚类(MC),在C57BL/6N模型中,它们通常表现出相似的调控趋势,其中 EC 2 和 4 与恶病质进展的关联最为明显且一致。在恶病质中活性显著增加的聚类2中的基序包括与炎症反应相关的转录因子识别的基序,例如信号转导和转录激活因子(STAT) 3和糖皮质激素受体(GR,由Nr3c1编码)(图2 E)。在恶病质中基序活性降低的转录因子包括肝细胞身份因子,例如核受体ONECUT1和NR1H3。
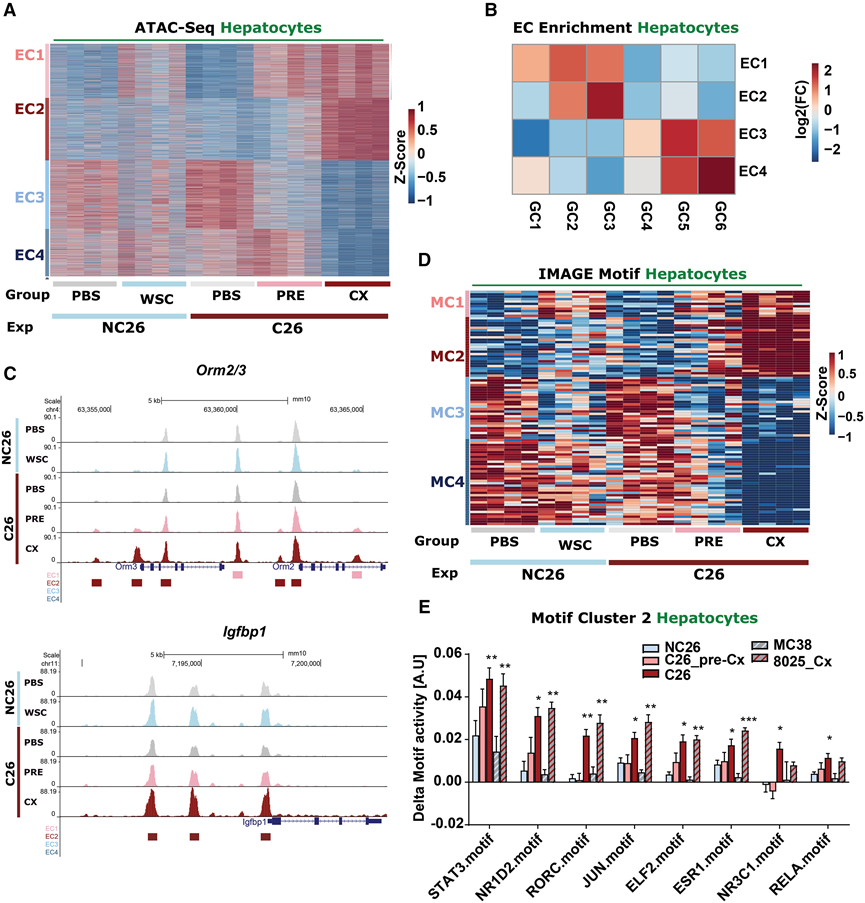
图2. 增强子和转录因子动力学驱动肝细胞对恶病质的反应。
(A) 对在一种或多种条件(BALB/c 模型)下差异可及的增强子进行行尺度 ATAC-seq 标签计数的 K 均值聚类。(B) 增强子聚类 (EC1–EC4) 的富集分析。(C) HEP-INTACT 小鼠 GFP+ 肝细胞核中口腔黏蛋白 2/3 (Orm2/3) 和胰岛素样生长因子结合蛋白 1 (Igfbp1) 位点的 ATAC-seq 数据。(D) 对 HEP-INTACT 小鼠 GFP+ 肝细胞核中,在一种或多种条件(BALB/c 模型)下差异活性转录因子基序的行尺度 IMAGE 预测活性。(E) 基序聚类 2 中预测具有恶病质诱导基序活性的前 8 个转录因子的基序活性平均变化。
03
恶病质促进肝细胞状态改变
值得注意的是,MC2 中得分最高的两个基序是 NR1D2 和 RORC(图 2 E)。它们与两个核激素受体家族相关,即 REV-ERB 和视黄酸孤儿相关受体 (ROR),它们是生物钟的关键成员,并与高度相似的基序结合(图 3 A)。生物钟以 24 小时周期性的昼夜节律方式调节其靶基因的表达,控制生理稳态。它由相互关联的转录和翻译反馈环路组成。转录激活因子脑和肌肉 ARNT-like 1(BMAL1,由Arntl1编码)的异二聚体和昼夜节律运动输出周期失效,驱动节律性基因表达。靶标包括核受体 REV-ERBα 和 β(分别由Nr1d1和Nr1d2编码),它们作为转录抑制因子发挥作用。REV-ERBα/β与ROR(由Rora、Rorb和Rorc编码)竞争启动子结合,并抑制Arntl1表达。因此,REV-ERB 形成一个负反馈回路,维持系统的节律性。
在这两个已鉴定的家族(ROR和REV-ERB)中的五种转录因子中,在恶病质小鼠中观察到基因表达变化最剧烈的是Nr1d1 (编码REV-ERBα),其次是Nr1d2(编码REV-ERBβ)(图3 B)。在C26和8025荷瘤小鼠的肝细胞核中,REV-ERBα的蛋白表达水平降低。REV-ERBα的表达在接近授时因子时间(ZT)9(即从研究动物中采集样本的时间点)时出现昼夜峰值。
为了进一步探索REV-ERB在恶病质期间肝细胞基因调控中的潜在作用,他们将肝细胞特异性REV-ERBα和REB-ERBβ缺陷的健康小鼠的公开RNA-seq数据整合到分析中。IMAGE预测的NR1D2/RORC基序靶基因在RNA-seq数据的GC3区高度富集(图3C)。在ZT10时,肝脏REV-ERBα和REB-ERBβ 29缺陷的小鼠中,该聚类(GC3)中的基因优先发生诱导表达(图3D)。进一步分析表明,其他核心时钟成员[如 D-box 结合 PAR 结构域碱性亮氨酸拉链转录因子 (DBP)]识别的基序也在恶病质进展过程中表现出差异基序活性的变化。他们观察到恶病质小鼠中Dbp的抑制,其昼夜峰值接近 ZT9(图 3 E)。相反,在恶病质动物中,在 ZT9 时诱导了昼夜峰值相反且为 REV-ERBα 靶标的时钟转录因子(如Arntl(编码 BMAL1)、Nfil3(编码核因子白细胞介素 3 调节)和Npas2(编码神经元 PAS 结构域蛋白 2)的表达(图 3 E)。
综上所述,这些结果表明,REV-ERB 可作为 GC3 基因表达的分子断裂。这些转录因子表达降低可能有助于恶病质进展过程中 GC3 基因的激活。
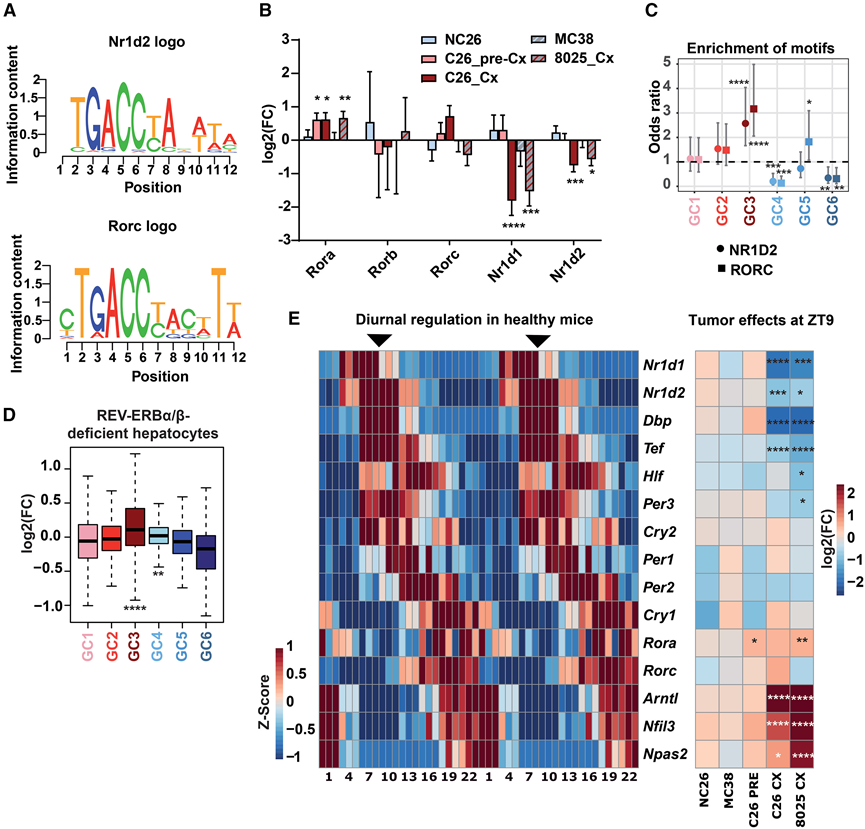
图3. 恶病质促进肝细胞核心时钟的改变。
(A) 恶病质激活基序的 NR1D2 和 RORC 基序的序列标识。(B) ROR 和 NR1D 家族中转录因子基因的表达变化。(C) 基因聚类(GC1-GC6) 内 NR1D2 和 RORC 基序的 IMAGE 预测靶基因的富集。(D) 六个肝细胞 GC 中肝细胞特异性 REV-ERBα/ꞵ 基因缺乏引起的表达变化。(E) 热图显示健康小鼠核心生物钟基因的昼夜表达(左图)以及这些基因在所示肿瘤模型中的表达变化(右图)。
04
恢复C26肿瘤小鼠肝脏REV-ERBα表达可改善恶病质特征
肝细胞中Nr1d1表达下降可能是驱动转录变化的关键因素,因此他们检测了REV-ERBα过表达对CCx发展的影响。通过肝细胞特异性甲状腺素结合球蛋白(TBG)启动子驱动的腺相关病毒(AAV),他们向BALB/c小鼠注射表达REV-ERBα或GFP(对照)的病毒载体,随后皮下移植C26肿瘤细胞(图4A)。肝脏蛋白表达检测证实,接受REV-ERBα AAV的小鼠肝脏中REV-ERBα表达升高,同时BMAL1表达降低。
REV-ERBα 过表达减轻了 C26 肿瘤动物的体重减轻(图 4B-C),并显著增加了心脏、胃肠道肌肉和 WAT 库的重量(图 4D-F)。两组之间的肝脏、脾脏和肿瘤样本重量相似(图 4G)。鉴于全身炎症在恶病质中的作用,他们接下来检测肝脏 REV-ERBα 过表达是否影响相关炎症介质。对照组和 REV-ERBα 过表达小鼠的循环中关键炎症恶病质介质(包括 IL-6、TNF-α、IL-1ꞵ 和 IL-22)水平相当(图 4H),这表明在 C26 肿瘤小鼠中观察到的 REV-ERBα 过表达的影响与整体全身炎症无关。然而,IL-27 已确定为 CCx 中肝脏转录反应的潜在上游调节因子(图 1 G),在肿瘤小鼠的肝脏过表达 REV-ERBα 后显著增加(图 4 I)。
由于在荷瘤小鼠中肝脏 REV-ERBα 过表达导致心脏重量增加,他们分析了其转录组作为导致 CCx 死亡的原型靶组织。该分析揭示了两种或多种情况之间 2560 个基因的差异表达,随后将其分为四个不同的心脏基因表达聚类(图 4 J)。值得注意的是,在第二个聚类中,其特征是在恶病质对照 C26 荷瘤小鼠中上调,而在 REV-ERBα 过表达时反应显著降低(图 4 J),“分解代谢过程的正向调节”是富集最强的通路之一(图 4 K)。此外,该聚类包含与自噬和巨自噬相关的通路。事实上,与 GFP 对照相比,在肝脏 REV-ERBα 过表达的 C26 肿瘤小鼠的心脏中,许多与自噬相关的基因的表达显著降低,包括微管相关蛋白 1 轻链 3β (Map1lc3b) 和 BCL2/腺病毒 E1B 相互作用蛋白 3 (Bnip3)。他们通过 qPCR 和蛋白质水平分析验证了Map1lc3b表达的降低,进一步表明诱导肝脏 REV-ERBα 表达可降低心脏的自噬并减少组织消耗(图 4L)。此外,在肝脏 REV-ERBα 过表达的 C26 肿瘤动物的心脏中,Trim63的 mRNA 水平降低。虽然在 GC 肌肉中未观察到Map1lc3b mRNA 表达的变化,但在肝脏 REV-ERBα 表达恢复的小鼠中,GC 肌肉重量增加与萎缩相关转录本Trim63和 F-Box 蛋白 32(Fbxo32)水平显著降低有关(图 4 M)。
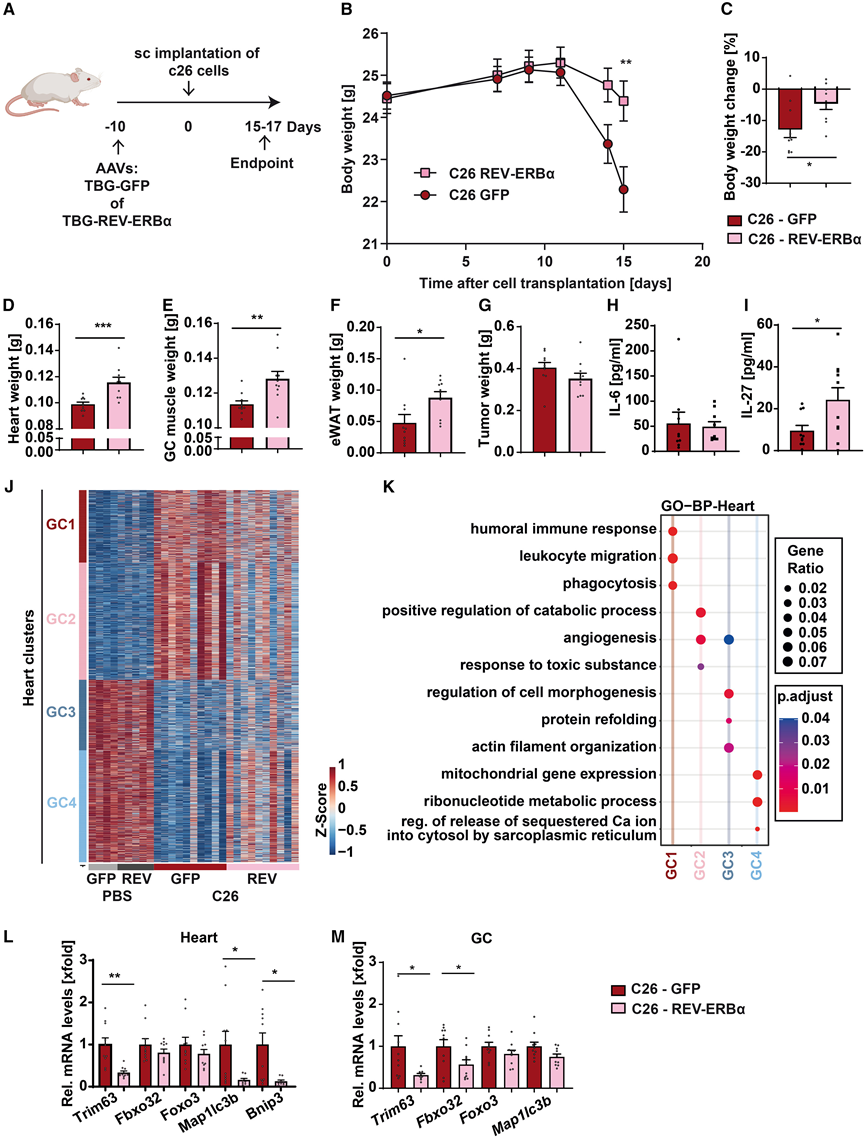
图4. 恢复C26肿瘤小鼠肝脏REV-ERBα表达可改善恶病质特征。
(A) 实验流程:给 BALB/c 小鼠注射 AAV 以实现肝细胞特异性 REV-ERBα 或 GFP(对照)表达,并在 10 天后皮下植入 C26 细胞。(B) C26 肿瘤小鼠的体重发展情况。(C-G) 体重变化以及心脏、腓肠肌、附睾白色脂肪组织(eWAT)和肿瘤的重量。(H-I) IL-6和 IL-27 的血清水平。(J) 心脏中差异表达基因分析。(K) 功能富集分析。(L-M)通过 qPCR 测定心脏和 GC 肌肉萎缩相关基因Trim63、Fbxo32和Foxo3以及自噬相关基因Map1lc3b和Bnip3 的mRNA 水平。
05
恶病质诱导的肝细胞分泌因子促进分解代谢过程
为鉴定REV-ERBα调控且可能介导CCx组织消耗的肝脏分泌因子,他们从GC3肝细胞CCx模型中筛选出13个编码分泌蛋白的候选基因(图1F),这些基因在C26和8025两种恶病质模型中均呈现≥8倍的稳定诱导。通过分析肝细胞特异性REV-ERBα/β缺陷小鼠肝脏数据,发现脂多糖结合蛋白(Lbp)、α-胰蛋白酶抑制剂重链H3(Itih3)、趋化因子CXCL14(Cxcl14)和胰岛素样生长因子结合蛋白1(Igfbp1)在一个或多个时间点较对照组显著上调(图5A-D)。相反,在肝脏REV-ERBα过表达转基因小鼠的公开数据中,Lbp、Itih3和Igfbp1在光照期表达显著降低。此外,已有肝脏REV-ERBα染色质免疫沉淀(ChIP)数据显示,REV-ERBα能与Lbp、Igfbp1和Itih3的启动子及邻近调控区结合,验证其作为这些肝细胞分泌因子转录抑制因子的作用。而Cxcl14的表达似乎与REV-ERBα基因组结合无明确关联。
接下来,他们评估了肝脏BMAL1缺陷小鼠中候选肝细胞分泌因子的表达。正如预期,肝脏Arntl(编码BMAL1)表达降低导致肝脏中Nr1d1(编码REV-ERBα)表达下调。值得注意的是,在BMAL1缺陷小鼠中,肝细胞分泌因子的mRNA水平和血液循环水平均升高,这与REV-ERBα敲低后观察到的模式相似。此外,在另外两种 CCx 模型:Lewis 肺癌 (LLC) 模型,其中 LLC 细胞经皮下移植,以及 Apc Min/+模型(一种结直肠癌遗传模型),观察到肝脏Nr1d1 mRNA 水平类似降低以及Lbp、Itih3和Igfbp1水平升高。这些发现表明 REV-ERBα 的下调和相关的肝细胞分泌因子分泌增加可能是不同模型中 CCx 的共同特征。
为了检测已鉴定的肝脏分泌因子是否能够在受恶病质影响的细胞类型中引发分解代谢反应,他们在体外用重组蛋白处理了原代大鼠心肌细胞、C2C12 肌管和分化的白色脂肪细胞(图 5 E)。在原代大鼠心肌细胞和 C2C12 肌管中,重组 LBP、ITIH3 和 IGFBP1 诱导心肌细胞大小(图 5 F)和肌管直径(图 5 G)显著且剂量依赖性地减小,表明存在萎缩效应。用重组 CXCL14 处理后没有观察到任何变化(图 5 F)。在分化的白色脂肪细胞中,重组 LBP 和 ITIH3 处理导致培养基中甘油释放增加,表明脂肪细胞中脂肪分解的诱导 (图 5 H)。相反,IGFBP1 和 CXCL14 处理不会导致培养基甘油水平显著增加(图 5 H)。 LBP、ITIH3 和 IGFBP1 联合处理即使在较低浓度下也会导致 C2C12 肌管出现萎缩效应(图 5 I)。用 C26 荷瘤小鼠的血浆处理 C2C12 肌管也会导致萎缩,而用抗肝细胞分泌因子中和抗体预处理血浆可消除这种效应,从而凸显了其治疗潜力(图5J-L)。
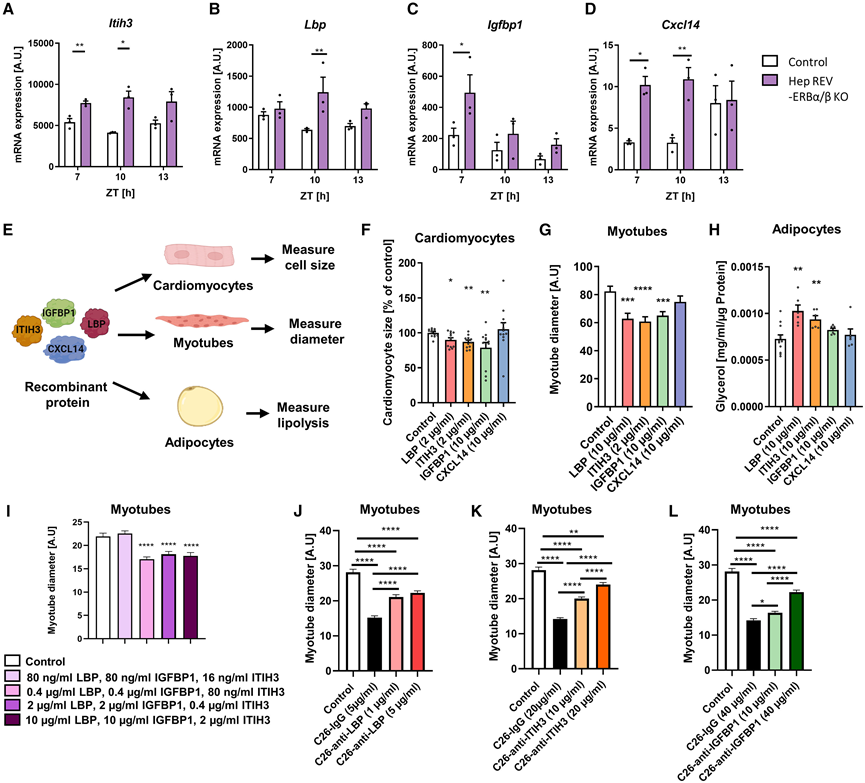
图5. 恶病质中时钟操纵后改变的肝细胞分泌因子在体外诱导分解代谢反应。
(A-D) 肝细胞特异性 REV-ERBα/β KO 小鼠的肝脏候选基因 mRNA 水平。(E) 在体外对原代大鼠心肌细胞、C2C12 肌管和分化的白色脂肪细胞进行肝脏候选重组蛋白测试的实验流程。(F) 将原代大鼠心肌细胞用指定剂量的 LBP、ITIH3、IGFBP1 或 CXCL14 处理 24 小时,然后用多聚甲醛 (PFA) 固定和分析。(G) 将 C2C12 肌管用指定剂量的 LBP、ITIH3、IGFBP1 或 CXCL14 处理 48 小时。使用 ImageJ 对孔进行成像并测量肌管直径。(H) 分化后的脂肪细胞用指定剂量的 LBP、ITIH3、IGFBP1 或 CXCL14 处理 24 小时。测量甘油含量并将其标准化为每孔的蛋白质含量。(I) 将 C2C12 肌管按所示用重组蛋白组合处理 48 小时,并使用 ImageJ 定量肌管直径。 (J-L) 用 PFA 固定细胞,成像,并使用 ImageJ 测量肌管直径。
06
肝细胞分泌因子敲低可改善体内癌症恶病质的特征
为了评估肝细胞分泌因子对体内CCx 的贡献,他们在 C26 恶病质模型中进行了肝脏 AAV 介导的Lbp (shLbp)、Itih3 (shItih3) 和Igfbp1 (shIgfbp1) 短发夹 RNA (shRNA) 敲低,单独或成对组合进行(图 6 A)。单独敲低Lbp和Itih3可降低 C26 荷瘤小鼠肝脏中它们各自的 mRNA 水平,但与对照 AAV (shControl) 相比,它并没有改变体重减轻或影响组织重量(图6B-E)。虽然Itih3敲低后没有观察到肌肉功能的改善,但Lbp敲低与握力增加相关,表明肌肉功能改善。
Lbp和Itih3 (shLbp-Itih3)的联合敲低通过肝脏 mRNA 表达的降低得到证实,这导致血浆蛋白水平显著降低(图6F),表明肝细胞是 CCx 循环中这些因子的主要来源。联合敲低导致 eWAT 重量增加,表明其对 C26 肿瘤小鼠的脂肪组织损失具有保护作用(图6H)。然而,整体体重、GC 肌肉、心脏、肝脏和肿瘤重量并未受到影响(图6G)。此外,他们观察到肌肉功能改善的趋势。肝脏Igfbp1 mRNA 水平显著降低(图 6 I)证实了Igfbp1的敲低,这也导致循环中 IGFBP1 水平显著降低(图 6 J),但不影响血浆 LBP 和 ITIH3 水平。值得注意的是,肝脏Igfbp1敲低导致 C26 荷瘤小鼠的体重减轻得到改善(图6K-L)。此外,肝脏 IGFBP1 敲低改善了 GC 肌肉重量、心脏重量和肌肉功能,而与用对照 AAV 治疗的 C26 荷瘤小鼠相比,脂肪组织、肝脏和肿瘤重量保持不变(图6M-O)。
他们观察到在REV-ERBα恢复的荷瘤小鼠肝脏中,Lbp和Itih3显著降低(图6 P)。同样地,在C26荷瘤小鼠中,LBP和ITIH3的循环水平显著升高,而在REV-ERBα过表达后,LBP和ITIH3的循环水平再次降低(图6Q-S)。在肝脏REV-ERBα过表达的恶病质C26荷瘤小鼠中,IGFBP1的蛋白质和mRNA水平与GFP对照小鼠相似(图6 P-T)。
综上所述,他们鉴定出REV-ERBα调控的肝细胞分泌因子,这些因子在CCx治疗期间循环中升高,并能够在体外诱导靶细胞的分解代谢过程。在肝脏中敲低这些肝细胞分泌因子可改变CCx在体内的特性,其中IGFBP1成为全身性肌肉萎缩的关键驱动因素。这些发现强调了肝脏通过分泌恶病质诱导的肝细胞分泌因子在促进CCx治疗期间组织萎缩中的作用。
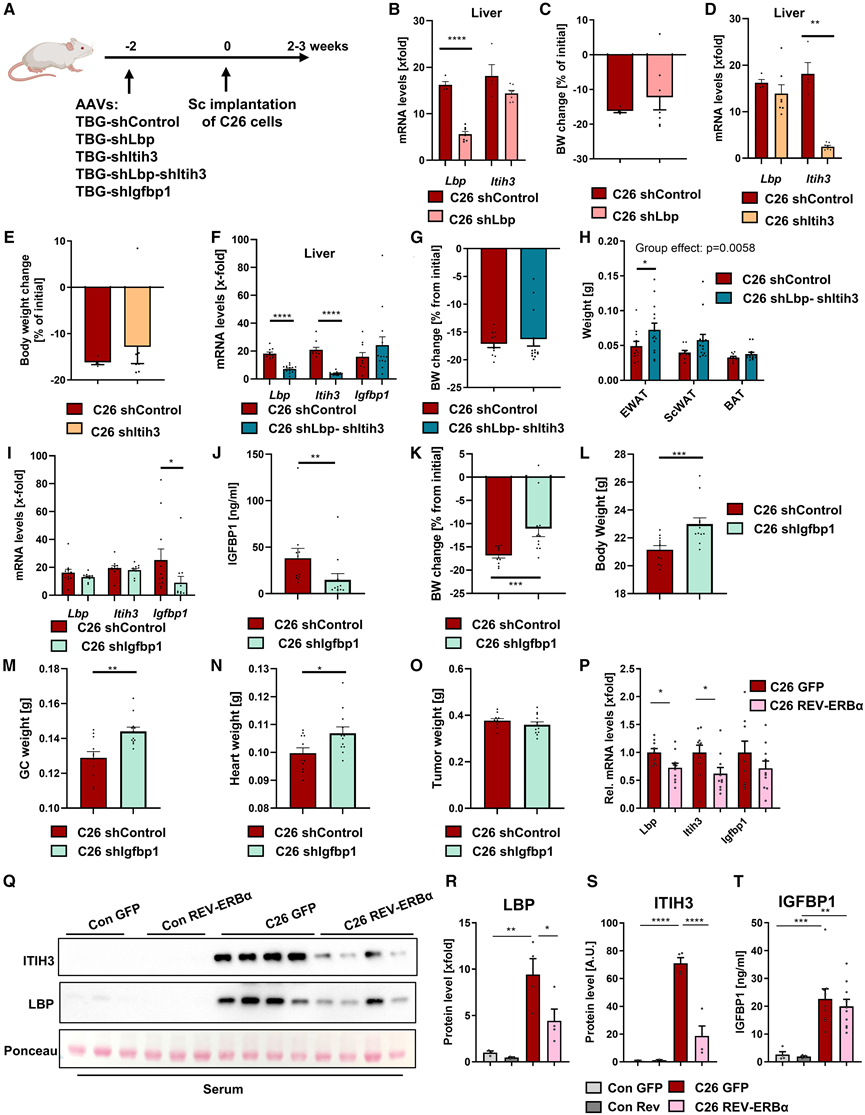
图6. 肝细胞分泌因子敲低可改善体内癌症恶病质的特征。
(A)实验流程。(B) 在 C26 肿瘤小鼠中,Lbp敲低后Lbp和Itih3的肝脏 mRNA 表达。(C) 体重变化分析。(D) C26 荷瘤小鼠中,敲低Itih3 基因后肝脏Lbp和Itih3 mRNA 的表达情况。(E)C26 肿瘤小鼠敲低Itih3后体重的变化。(F) 在 C26 肿瘤小鼠中,Lbp和Itih3联合敲低后肝脏中肝细胞分泌因子的 mRNA 表达。(G) 体重变化。(H) 脂肪组织重量。(I) 在 C26 肿瘤小鼠中,Igfbp1敲低后肝脏中肝细胞分泌因子的 mRNA 表达。(J) 通过 ELISA 测定循环中 IGFBP1 水平。(K) Igfbp1敲低后 C26 肿瘤小鼠体重的变化。(L-O) Igfbp1敲低后的最终身体和组织重量。(P) 肝脏候选基因 mRNA 水平。(Q-S) 血清中 ITIH3 和 LBP 的蛋白质表达及随后的定量。(T) 通过 ELISA 测定 C26 荷瘤小鼠血清中的 IGFBP1 水平。
07
恶病质癌症患者肝脏分泌因子增多
最后,他们评估了肝脏分泌因子在不同癌症类型恶病质患者中是否也增加。第一个队列由不同胃肠道癌症实体的患者(患者队列 1)组成,这些患者体重稳定或恶病质,以及健康对照者。恶病质癌症患者的平均体重变化为 -13.95%,而体重稳定的癌症患者体重变化为 -1.52%(图 7 A)。此外,恶病质癌症患者的循环 C 反应蛋白水平较高,而血红蛋白、白蛋白和葡萄糖水平较低。与小鼠数据一致,与健康对照者相比,恶病质癌症患者血浆中的 LBP、ITIH3 和 IGFBP1 蛋白水平显著升高(图7B-C)。此外,与体重稳定的癌症患者相比,恶病质癌症患者的 IGFBP1 蛋白显著升高(图 7 D)。值得注意的是,LBP、ITIH3 和 IGFBP1 的循环血浆蛋白水平与人类受试者报告的平均体重变化呈显著相关性(图7E-G)。患者队列 2 包括 24 名体重稳定和 44 名恶病质胰腺癌患者(图 7 H)。与之前的研究结果一致,与体重稳定的患者相比,恶病质患者的循环 LBP、ITIH3 和 IGFBP1 水平升高(图7I-K)。最后,他们评估了 72 名肺癌患者的肝细胞分泌因子水平,细分为 49 名体重稳定个体和 23 名恶病质个体(患者队列 3)(图 7 L),使用靶向蛋白质组学方法分析血浆样本。与患者队列 1 和 2 相似,与体重稳定的癌症患者相比,LBP、ITIH3 和 IGFBP1 蛋白表达显著增加,并且 LBP 和 ITIH3 水平与体重减轻显著相关(图7M-O)。
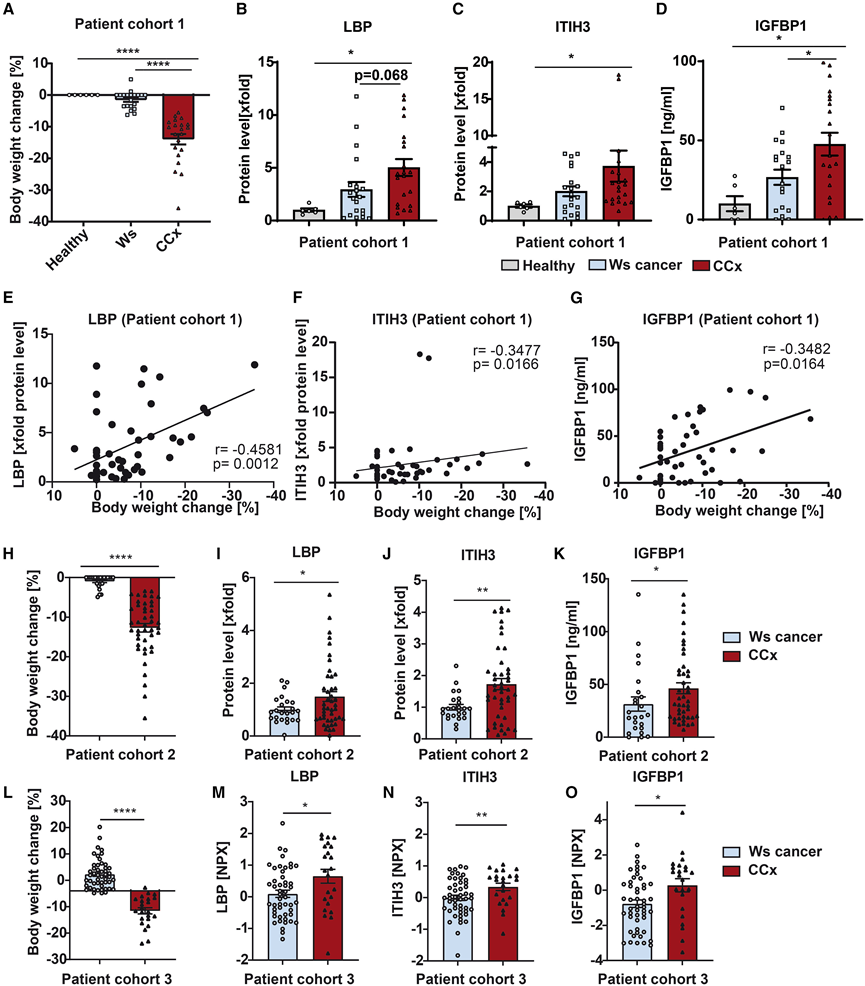
图7. 恶病质癌症患者肝细胞分泌因子增多。
(A) 健康人类、体重稳定和恶病质胃肠道癌症患者队列的相对体重变化。(B-C) 对患者队列 1 中的 LBP和 ITIH3循环蛋白水平进行定量。(D) 通过 ELISA确定患者队列 1 中个体的 IGFBP1 循环水平。(E-G)LBP、ITIH3和IGFBP1的循环水平与患者队列 1 的体重变化的相关性。(H) 体重稳定和恶病质胰腺癌患者队列的相对体重变化。(I-J) 循环蛋白水平的定量。(K) 通过 ELISA确定患者队列 2 的 IGFBP1 循环水平。(L) 体重稳定和恶病质肺癌患者队列的相对体重变化。(M-O) 通过靶向蛋白质组学方法分析血浆中LBP、ITIH3和IGFBP1的标准化蛋白表达。
+ + + + + + + + + + +
结 论
本研究对各种体重稳定的癌症和癌症恶病质模型中的肝脏进行了转录组学和基因组学分析。整合的多层次分析方法确定了一个独特的基因表达特征,包括肝细胞分泌因子和生物钟成分REV-ERBα,它们是癌症恶病质中肝脏转录重编程的关键调节因子。值得注意的是,恶病质中肝细胞特异性REV-ERBα的基因重建改善了外周组织消耗。这种改善与特定恶病质控制的肝细胞分泌因子水平降低相关。这些肝细胞分泌因子促进多种细胞类型的分解代谢,并且在恶病质癌症患者中水平升高。本项研究结果揭示了肝脏在癌症恶病质中导致外周组织消耗的机制,为未来的治疗干预提供了前景。
+ + + + +



