

 English
English文献解读|Cell Rep Med(10.6):对原发性干燥综合征患者肠道微生物组和代谢组的多方面特征分析
✦ +
+
论文ID
原名:Multi-faceted characterization of the gut microbiome and metabolome in patients with primary Sjögren syndrome
译名:对原发性干燥综合征患者肠道微生物组和代谢组的多方面特征分析
期刊:Cell Reports Medicine
影响因子:10.6
发表时间:2026.5.19
DOI号:10.1016/j.xcrm.2026.102777
背 景
原发性干燥综合征(pSS)是一种慢性系统性自身免疫性疾病,其特征是外分泌腺的炎症和破坏,临床表现为眼干和口干(干燥性角结膜炎和口干症)。pSS的发病机制涉及针对自身抗原的异常免疫反应,导致上皮细胞损伤和唾液腺及泪腺分泌功能障碍。除腺体受累外,pSS 患者还可能出现多种系统性表现,包括关节炎、间质性肺病(ILD)、肾脏受累、神经系统并发症以及淋巴瘤风险增加。虽然自身免疫性上皮炎的概念已提出用于解释 pSS 的发病机制,但其确切的潜在机制仍未完全阐明。遗传易感性和环境因素均与该病的发生和发展有关。
实验设计
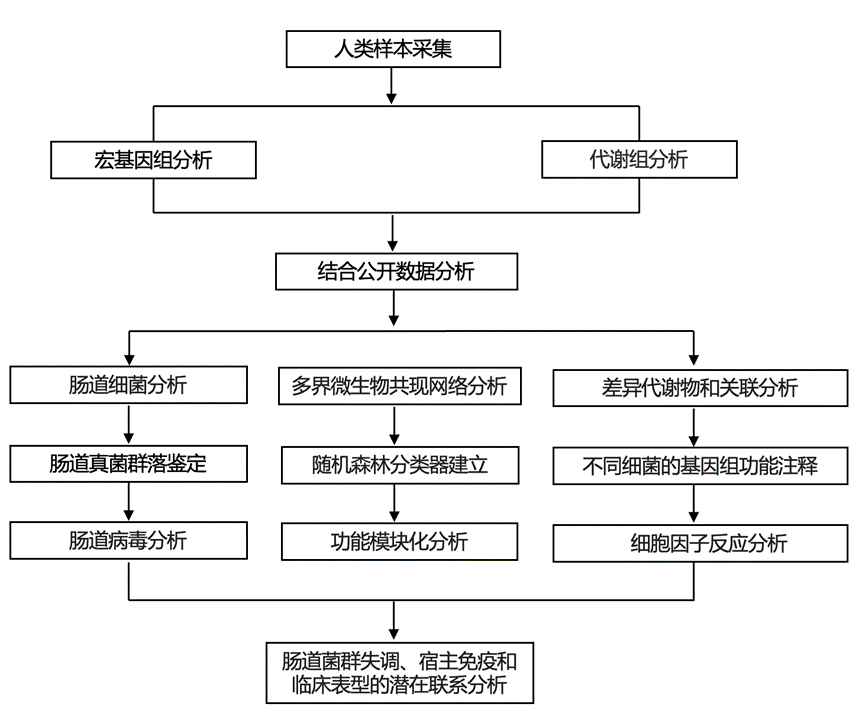
结 果
01

原发性干燥综合征患者肠道多界微生物组的多样性和组成
为了研究pSS患者的肠道微生物组和代谢组,研究团队收集了206例pSS患者和355例非pSS对照组的粪便样本。研究人群分析显示,pSS患者的年龄略高于非pSS对照组(平均年龄57.5岁 vs. 53.0岁),且女性比例显著高于对照组。在pSS患者中,68.9%有药物使用史,包括泼尼松、来氟米特、羟氯喹和吗替麦考酚酯等药物。此外,16.0%的pSS患者诊断患有间质性肺病(ILD),进一步凸显了该合并症在pSS人群中的高发率。他们对206例pSS患者和355例非pSS受试者的粪便样本进行了全宏基因组测序。稀疏曲线分析显示,随着样本量的增加,非pSS受试者的细菌丰富度略高于pSS患者(图1A)。然而,两组之间的物种丰富度和香农多样性均无显著差异(图1B-C),表明pSS对肠道细菌α多样性的影响有限。在校正药物使用后,基于Bray-Curtis距离的主坐标分析(PCoA)仍然显示pSS患者和非pSS受试者之间存在显著差异(图1D),表明肠道菌群组成的变化独立于药物治疗而持续存在,并且与pSS相关。
在门水平上,厚壁菌门、拟杆菌门、放线菌门和变形菌门在两组中均占主导地位(图 1 E)。pSS患者的变形菌门丰度显著高于对照组,而拟杆菌门丰度则较低。在属水平上,pSS 患者表现出大肠杆菌属、链球菌属、韦荣氏球菌属、轻乳杆菌属和局限乳菌杆属以及其他 17 个属的丰度升高,而粪杆菌属、拟杆菌相关菌属、拟杆菌属、罗斯氏菌属和厌氧丁酸菌属以及其他11个属在非 pSS 对照组中富集(图 1 F)。
在物种水平上,49个分类单元在各组间存在显著差异,其中19个物种在pSS患者中富集,30个物种在对照组中富集(图1H)。韦荣氏球菌目(例如,小韦荣氏球菌)、乳杆菌目(例如,粘膜乳杆菌、唾液乳杆菌和副溶血链球菌)以及肠杆菌目(例如,大肠杆菌、肺炎克雷伯菌和弗氏柠檬酸杆菌)的物种在pSS患者中显著增加。相反,一些拟杆菌门成员,例如Prevotella copri clade A、Phocaeicola vulgatus、Bacteroidota uniformis、Anaerostipes hadrus, 和Phocaeicola plebeius,在pSS患者中显著减少。在校正性别、年龄、体重指数(BMI)和用药情况后,显著物种的数量略有减少,但大多数差异性分类群,包括副溶血链球菌、唾液乳杆菌和微小韦荣氏球菌,仍然与pSS密切相关。
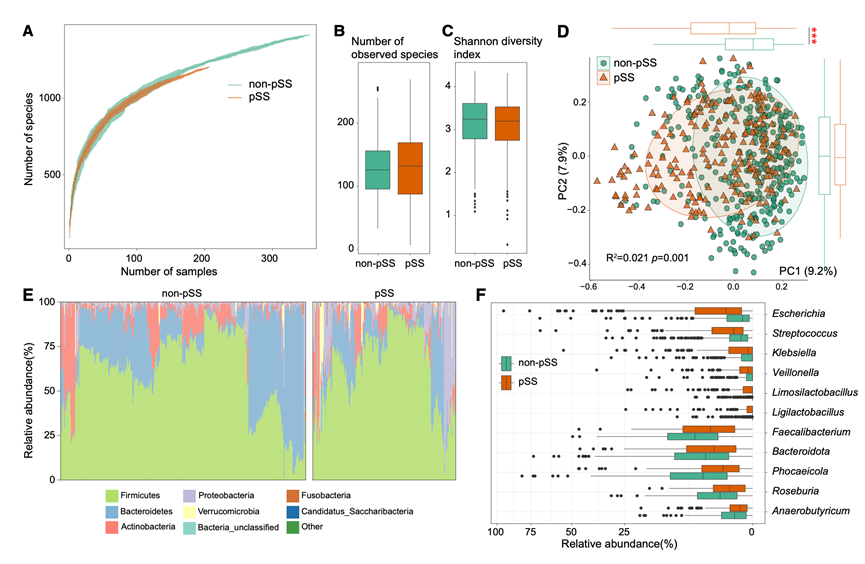
图1. 原发性干燥综合征 (pSS) 患者与非 pSS 对照组的肠道菌群多样性和组成差异。
(A) 稀疏曲线描绘了 pSS(橙色)和非 pSS(绿色)组中观察到的细菌物种的累积数量与测序深度的关系。(B-C) 箱线图比较 pSS 组和非 pSS 组之间观察到的细菌物种数量和香农多样性指数。(D) 基于 Bray-Curtis 距离的物种水平主坐标分析 (PCoA)。(E) 所有样本中排名前八的细菌门的相对丰度。(F) 箱线图显示组间显著改变的属。
02
肠道真菌群落的多样性和组成
稀疏曲线分析显示,非pSS受试者的真菌总丰富度显著高于pSS患者(图2A)。相反,非pSS个体中观察到的物种的样本间离散度较低,表明其真菌群落结构比pSS患者更为均一(图2B)。香农多样性指数在两组间也存在显著差异(图2C)。校正药物使用后,基于Bray-Curtis主坐标分析(PCoA)显示,pSS患者和非pSS对照组的肠道真菌群落存在明显的聚类差异(图2D),这与细菌群落中观察到的菌群失调相一致。
从组成上看,所有受试者的肠道真菌群主要由酵母菌(Saccharomycotina)构成,其次是Pezizomycotina、担子菌(Basidiomycota)和毛霉菌(Mucoromycota)(图2E)。在属水平上,念珠菌属(Candida)、科达酵母属(Malassezia)和圆盘菌属(Orbilia)在pSS患者中显著富集,而毕赤酵母(Pichia)、曲霉属(Aspergillus)和科达达酵母属(Kodamaea)在对照组中更为丰富(图2F)。共有19种真菌符合差异丰度标准,其中大多数在pSS患者中富集。具体而言,毕赤酵母(例Pichia Sumatraense和Pichia glabrum)、念珠菌(例如白色念珠菌)和马拉色菌属(例如Malassezia arunalokei和Malassezia restricta)的菌种在pSS患者中更为丰富。相反,仅有三种菌种,包括奥氏科达马菌、Rhizopus stolonifer和Aspergillus sp. c38,在患者中显著减少,提示它们可能作为该疾病的真菌生物标志物。在校正潜在混杂因素后,大多数真菌类群仍与pSS显著相关(图2G)。这些结果表明,真菌组成的变化,特别是念珠菌和马拉色菌种的富集,可能代表与pSS相关的微生物特征。
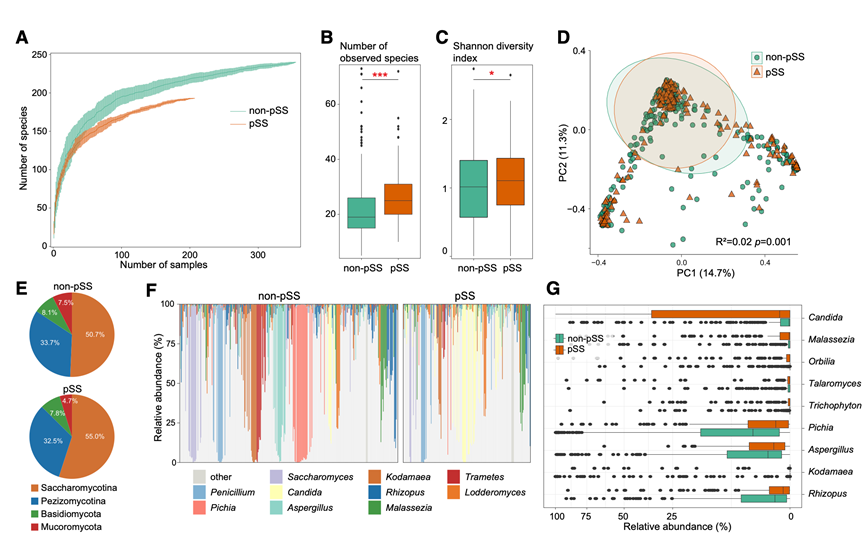
图2. 原发性干燥综合征 (pSS) 患者与非 pSS 对照组肠道真菌群落多样性及组成差异。
(A) 稀疏曲线描绘了 pSS(橙色)和非 pSS(绿色)组中观察到的真菌物种的累积数量与测序深度的关系。(B-C) 箱线图比较了 pSS 组和非 pSS 组中观察到的真菌物种数量和香农多样性指数。(D) 基于真菌物种水平的 Bray-Curtis 距离进行主坐标分析 (PCoA)。(E) 饼图显示了 pSS 患者和非 pSS 对照组的门级真菌群组成。(F) 所有样本中排名前 10 的真菌属的相对丰度。(G) 箱线图显示各组之间真菌属的显著变化。
03
肠道病毒组的多样性和组成
在561个宏基因组样本中,共定量了62816个病毒操作分类单元(vOTU),其中62.7%分配到33个已知的病毒科,用于后续分析。稀疏曲线分析显示,与非pSS患者相比,pSS患者的病毒丰富度略低(图3A),这一趋势与观察到的物种计数结果相符(图3B)。pSS患者的组内香农多样性也显著降低(图3C)。药物调整后,基于PCoA分析显示pSS组和非pSS组之间存在明显分离(图3D),表明肠道病毒群落结构存在持续性改变,且这种改变与药物作用无关。
接下来,他们从科水平分析了肠道病毒组的组成,并鉴定出Siphoviridae、Myoviridae、Podoviridae_crAss-like和Quimbyviridae为优势科(图3E)。值得注意的是,Myoviridae和Podoviridae在pSS患者中富集,而Quimbyviridae在非pSS受试者中显著升高。在vOTU水平上,1323个vOTU在pSS患者中表现出显著变化,其中327个富集,951个减少。在校正潜在混杂因素后,显著vOTU的数量显著减少,表明某些病毒关联部分受到个体特征的影响。对vOTU的宿主预测显示,在pSS患者中富集的病毒主要与大肠杆菌属、链球菌属、阿克曼菌属、克雷伯氏菌属和巨单胞菌属等细菌相关(图3F-G)。相反,在pSS患者中富集的vOTU主要与拟杆菌属、粪杆菌属、普雷沃氏菌属、副拟杆菌属和罗斯氏菌属相关。这些显著差异提示病毒可能在pSS的发病机制或疾病进展中发挥重要作用。
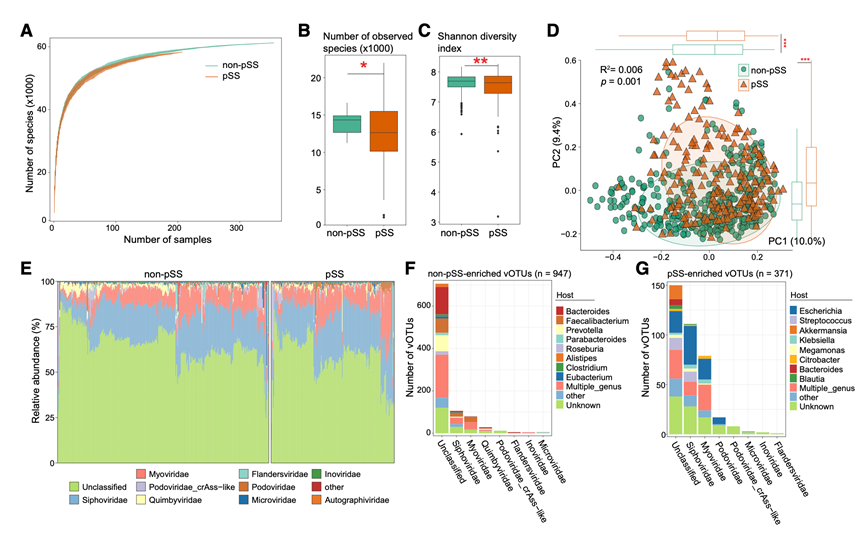
图3. pSS患者与非 pSS 对照组的肠道病毒组多样性和组成差异。
(A) 稀疏曲线描绘了 pSS(橙色)和非 pSS(绿色)组中观察到的病毒种类的累积数量与测序深度的关系。(B-C) 箱线图比较 pSS 组和非 pSS 组之间观察到的 vOTU 数量和香农多样性指数。(D) 基于VOTU水平的Bray-Curtis距离的PCoA分析。(E) 所有样本中排名前 10 的病毒家族的相对丰度。(F-G) 堆叠条形图显示了检测到的病毒种类及其预测的细菌宿主的分布情况。
04
肠道多界微生物共现网络
鉴于肠道生态系统中细菌、真菌和病毒的共存及其潜在的相互作用,以及患者和对照组之间这些微生物成分的显著差异,他们接下来研究了跨界相互作用网络。差异丰度多界特征(即49种细菌、19种真菌和1323个vOTU)的系统发育分布和富集模式总结在分类树中(图4A)。为了构建全面的相互作用网络,他们对这些特征进行了相关性分析,仅保留了显著相关的特征。最终得到的pSS患者和非pSS对照组的网络分别包含1160条边和1101条边(图4B-C)。在两个网络中,B. uniformis、粪杆菌A分支P. copri clade A、大肠杆菌和P. vulgatus均表现出较高的连接性,表明它们在肠道稳态中可能发挥关键作用(图4D-E)。他们进一步鉴定出116个物种,它们的相互作用模式在两个网络中存在显著差异。值得注意的是,Rothia mucilaginosa和Rhizopus stolonifer c238在pSS网络中表现出更高的连接性,这可能与疾病的发病机制有关。
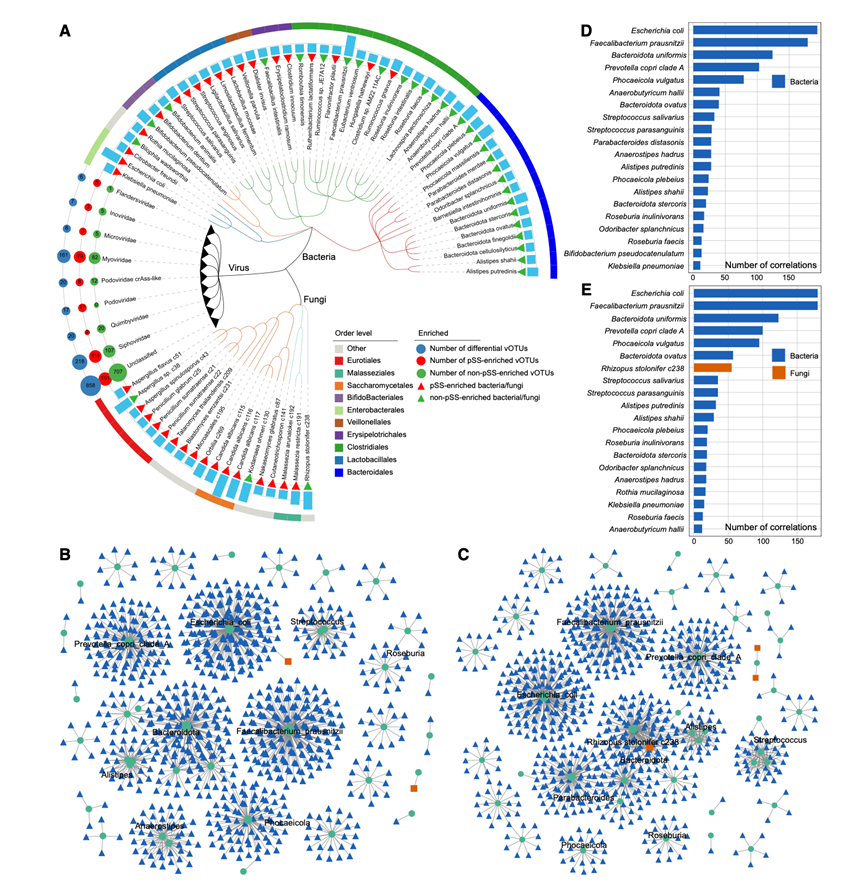
图4. 肠道微生物组的多界差异分类单元和共现网络。
(A) 系统发育树展示了pSS组和非pSS组之间显著改变的多界分类单元。(B-C) 非 pSS 组和 pSS 组中细菌、真菌和病毒类群的共现网络。(D-E) 柱状图显示了非 pSS 组和 pSS 组中连通性(度)最高的 20 个分类单元。
05
基于多界特征的疾病状态分类
为了评估肠道微生物谱能否区分pSS患者和健康对照组,他们分别使用细菌、真菌和病毒特征训练了随机森林分类器。基于29个细菌特征、10个真菌特征和64个病毒特征的模型分别达到了最佳曲线下面积(AUC)0.841、0.849和0.943(图5A)。值得注意的是,整合了这三个界(细菌、真菌和病毒)的联合模型优于单个分类器,其交叉验证AUC达到了0.950。排名靠前的特征包括pSS富集细菌,例如S. parasanguinis、Clostridium innocuum、Ruthenibacterium lactatiformans、L. salivarius和Streptococcus anginosus,以及一些vOTU(图5B-E)。为了验证这些特征的稳健性和普适性,他们从中国西部一家医院招募了一个独立的队列(56例原发性干燥综合征患者和58例健康对照者),该队列在地理位置上与东北地区的发现队列不同。他们观察到一致的分类性能,AUC值分别为0.711(细菌)、0.659(真菌)、0.730(病毒)和0.742(联合模型)(图5F)。包括Veillonella、Limosilactobacillus、Ligilactobacillus、Faecalibacterium、Escherichia和Candida在内的多个分类群在两个队列的原发性干燥综合征患者中均表现出可重复的富集趋势(图5G-J)。值得注意的是,在原发性干燥综合征患者中富集的菌种,例如V. parvula、S. parasanguinis和L. salivarius,在验证队列中也表现出一致的方向性变化。
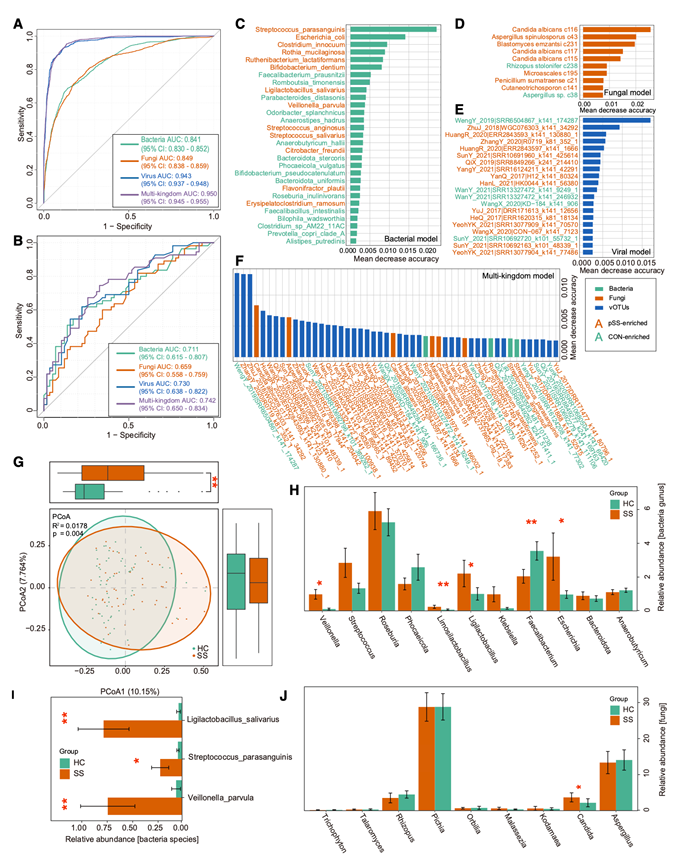
图5. 根据肠道多界特征组成对 pSS 状态进行分类。
(A) 分别使用细菌、真菌和病毒特征以及组合的多界特征对 pSS 进行分类的受试者工作特征(ROC)曲线。(B) 来自不同地区的独立队列的外部验证,显示细菌、真菌、病毒和多界组合模型的 ROC 曲线。(C-F) 对细菌、真菌、病毒和多界组合分类模型贡献最大的微生物特征。(G) 验证队列的 PCoA,显示 pSS 患者和非 pSS 对照组之间存在明显的聚类。(H-J) 先前在发现队列中鉴定为具有显著性的关键细菌属、细菌种和真菌属的相对丰度。
06
肠道菌群的功能特征
为了表征pSS患者肠道菌群的功能属性,他们应用UHGP流程对所有粪便宏基因组进行功能分析,获得了9041个同源基因簇(KO)和765个KEGG模块用于后续分析。功能多样性分析显示,与对照组相比,pSS患者的模块丰富度较低,但香农指数较高(图6A-B),表明不同样本间功能分布更为均匀。与分类学变化一致,模块组成的PCoA分析显示各组之间清晰分离(图6C),强调了pSS患者肠道菌群的显著功能重塑。在765个模块中,有120个模块在各组间存在显著差异,其中115个在pSS患者中富集,而只有5个在非pSS患者中富集(图6D-E)。pSS富集的模块主要参与芳香族化合物降解(例如苯甲酸、甲苯、二甲苯、邻氨基苯甲酸和酪氨酸)、多胺生物合成、细菌和真菌次级代谢产物生物合成、致病性、抗菌素耐药性和双组分调控系统。代表性例子包括葡萄球菌毒力调控系统SaeS-SaeR、锰/锌/铁转运系统和多重耐药模块。这些发现表明,pSS肠道微生物群的功能已发生重编程,增强了其应激反应、毒力潜能和外源性物质代谢能力。
为了从实验上评估肠道菌群失调的功能性后果,他们对75例pSS患者和66例非pSS对照组的配对血清和粪便样本进行了非靶向代谢组学分析。共有18种血清代谢物和7种粪便代谢物在两组间显示出显著差异(图6H-I)。在血清中,pSS患者表现出与氧化应激和免疫激活相关的代谢物水平升高,包括肌酐、吲哚硫酸盐、三甲胺N-氧化物(TMAO)、马尿酸、犬尿氨酸、色氨酸和尿酸——这与先前报道的自身免疫性疾病中的代谢改变一致。在粪便中,pSS患者的短链脂肪酸(SCFA)丁酸和丙酸的浓度显著降低,这与其他炎症性疾病中观察到的趋势相似。此外,吲哚(一种微生物色氨酸代谢产物和芳烃受体 (AhR) 配体,具有免疫调节特性)显著减少,提示pSS患者肠道环境中 AhR 介导的黏膜免疫调节功能受损。总的来说,这些代谢组学改变表明,系统性代谢状态向促炎和氧化状态转变,同时保护性微生物代谢产物减少,这可能反映了肠道菌群失调的功能性后果及其对宿主免疫稳态的影响。
为了探究微生物类群改变与其代谢功能之间的功能联系,他们构建了整合差异细菌物种和KEGG模块的相关网络,分别针对pSS组和非pSS组。鉴于模块均基于相同的宏基因组数据推断,他们预期其与细菌类群的关联性较强;因此,他们应用了严格的阈值以仅保留最稳健的相互作用(图6F-G)。在pSS网络中,大肠杆菌、肺炎克雷伯菌、副溶血链球菌和唾液链球菌表现出最高的连接性,表明它们在菌群失调的生态系统中发挥着核心作用。值得注意的是,这些物种与pSS富集的、参与毒力调控(例如SaeS-SaeR双组分系统)、金属铁转运和应激适应的模块密切相关——这些功能可能增强细菌在炎症条件下的持续存在。相比之下,非 pSS 网络以共生分类群(如F. prausnitzii、Alistipes putredinis和O doribacter splanchnicus)与 SCFA 生物合成和氨基酸代谢相关的模块之间的紧密联系为特征,这与稳态代谢特征一致。
为了将此分析扩展到细菌、真菌和血清/粪便代谢物之间的跨界关联,他们采用了更为宽松的阈值,以捕捉具有生物学意义的关联,因为域间关系本身就较弱(图 6J)。由此得到的整合网络揭示了与pSS相关的独特模式。几种富集于pSS 的细菌,包括S. anginosus、L. salivarius、Bifidobacterium dentium、S. parasanguinis、大肠杆菌和V. parvula,与SCFA呈显著负相关,而与促炎或毒性代谢物(例如,TMAO、吲哚硫酸盐、尿酸和犬尿氨酸)呈正相关。相反,一些有益的菌群,例如动物双歧杆菌和肠道粪杆菌,则呈现出相反的趋势,它们与短链脂肪酸呈正相关,与促炎代谢物呈负相关。大多数真菌类群,特别是三种在pSS中富集的白色念珠菌亚种,与有害代谢物表现出强烈的正相关性,这表明真菌菌群失调可能通过代谢串扰进一步加剧全身炎症。总而言之,这些结果揭示了pSS中复杂的、跨界的代谢重编程,其特征在于连接菌群失调细菌、真菌和促炎代谢物的协同致病网络,这些网络可能导致疾病的发生。
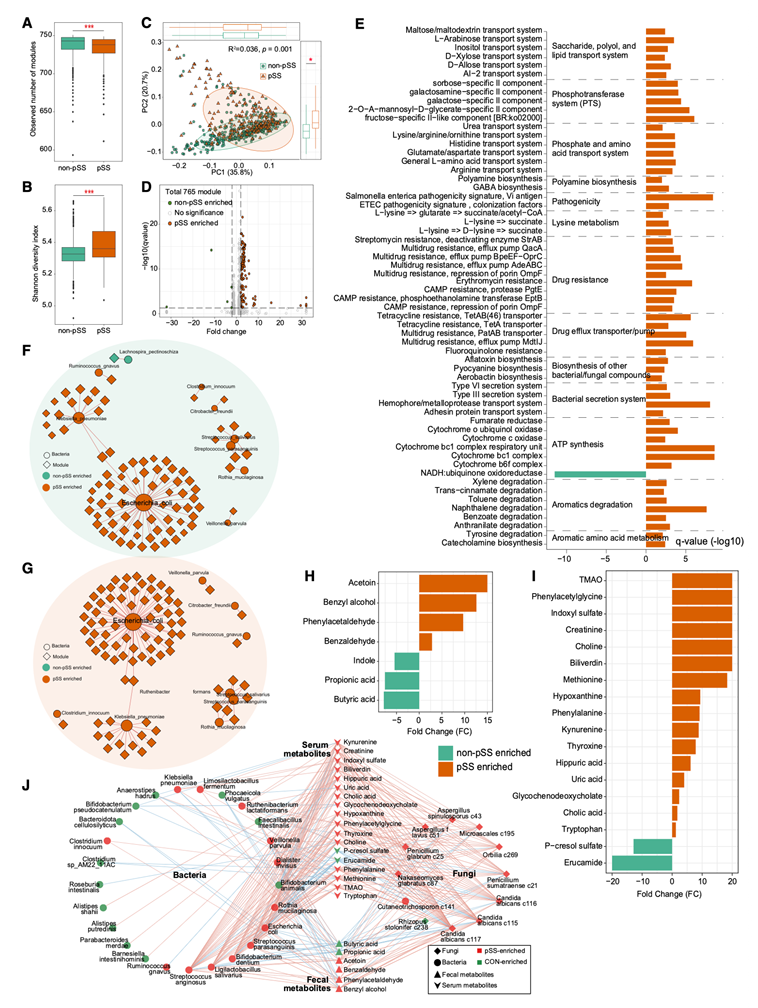
图6. 原发性干燥综合征患者的功能和代谢组学改变。
(A-B) 箱线图比较 pSS 组和非 pSS 组的功能模块丰富度和香农多样性指数。(C) 基于 Bray-Curtis 距离的主坐标分析(PCoA)显示各组间功能模块组成的总体差异。(D) 火山图突出显示了 pSS 组和非 pSS 组之间差异丰富的功能模块。(E) 条形图显示显著改变的功能模块,并标注了更高级别的功能分类和富集趋势。(F-G) 网络图描绘了差异丰度细菌/真菌分类群与功能模块之间的关联。(H-I) 柱状图显示通过非靶向代谢组学分析鉴定的粪便和血清中差异丰度的代谢物。(J) 将丰度不同的细菌和真菌类群与改变的粪便代谢物联系起来的相关网络。
07
基于MAG的肠道物种功能表征
为了进一步探索富含pSS的微生物的功能潜力,他们重建了三种关键物种(S. parasanguinis、L. salivarius、和V. parvula)的高质量宏基因组组装基因组(MAG),每种物种分别获得13、15和10个MAG。利用多个数据库对蛋白质编码基因进行了注释,包括B细胞和T细胞表位数据库、VFDB数据库、DRG(耐药基因数据库)以及抗菌肽预测工具。B细胞和T细胞表位预测均表明,所有物种都含有大量免疫原性蛋白(图7A-C)。保守的应激相关和代谢管家蛋白,例如BiP / GRP78、HSP60 / HSP70、α/β-烯醇化酶、磷酸甘油酸变位酶和丙酮酸激酶,在不同物种中均鉴定为抗原来源。每个物种也表现出独特的抗原谱。S. parasanguinis编码的表位来源于CTP合成酶、氨肽酶N和多种转录相关蛋白。V. parvula携带类似于胶原α链和促甲状腺激素受体的表位。L. salivarius含有多个与宿主线粒体和代谢蛋白同源的表位。一些表位与人类蛋白(例如胶原蛋白、PKM、IMPDH和CTPS)具有高度序列相似性,这提示这些微生物抗原可能通过分子模拟触发自身免疫反应。
其次,全面的毒力因子注释揭示了三种菌株之间不同的致病性状特征(图7A-C)。S. parasanguinis和L. salivarius拥有最高的毒力相关基因多样性,其中唾液乳杆菌编码多种与黏膜定植和免疫逃逸相关的元件,包括分选酶(srtC)、19种荚膜生物合成酶(cap8P、cpsA-I)和胆盐水解酶(bsh)。此外,还检测到了分子伴侣蛋白(DnaK、GroEL和ClpC/P/E)、糖基转移酶和双组分调控系统(RegX3和LisR),表明其具有增强的适应性和应激耐受能力。S. parasanguinis含有多种黏附因子和免疫相互作用因子,例如纤连蛋白结合蛋白(fbp54)、层粘连蛋白结合蛋白(lmb)和胶原结合黏附素(cnm)。此外,还鉴定出一些分泌相关的毒力决定因子,包括VII型分泌蛋白EsxA、金属蛋白酶ZmpB和黏附蛋白Lap,这些因子可能促进上皮细胞黏附并增强炎症反应。虽然V. parvula编码的典型毒力因子较少,但它含有应激和氧化还原相关蛋白(过氧化氢酶katA和硫氧还蛋白/蛋氨酸亚砜还原酶msrA / B)、黏附蛋白(Lap和LirB)以及荚膜多糖生物合成基因聚类(cps4I、ugd、galU和galE),这些蛋白可能有助于其在炎症条件下持续存在并调节免疫反应。
第三,宏基因组分析揭示了三种细菌中广泛存在的抗菌药物耐药性决定聚类,涵盖了固有耐药性和获得性耐药性(图7A-C)。S. parasanguinis携带多重耐药基因,使其对大环内酯类、林可酰胺类、利福平、四环素类和喹诺酮类药物产生耐药性。L. salivarius含有与氨基糖苷类、大环内酯类和四环素类耐药性相关的基因,而V. parvula主要表现出四环素耐药性和天然耐药性标记。相比之下,三种细菌中抗菌肽相关序列均较少,表明其产生抗菌化合物的固有能力相对较弱。
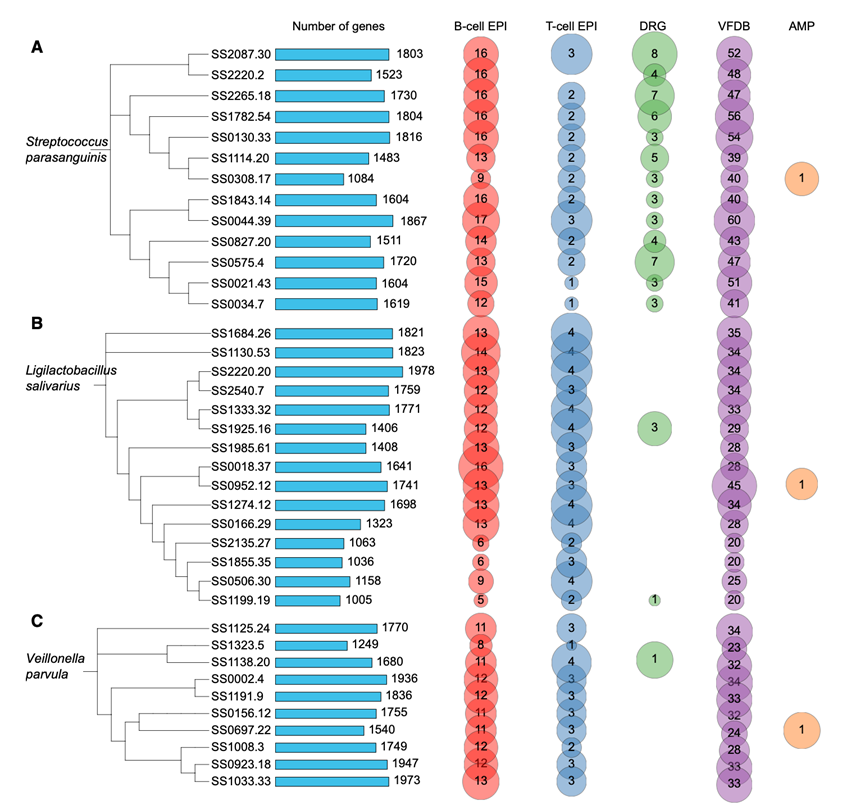
图7. 三种关键 pSS 相关物种的基因组特征。
(A-C) 不同细菌的MAG功能注释。
08
pSS相关肠道微生物诱导的免疫激活
多界肠道微生物组分析显示,pSS相关微生物群中的细菌(尤其是S. parasanguinis、V. parvula、L. salivarius)和真菌(特别是白色念珠菌)均具有可能破坏免疫稳态的分子特征。基于这些观察结果,他们利用外周血单核细胞(PBMC)进行了体外实验,以评估其免疫刺激潜力。将PBMC暴露于pSS患者的粪便微生物群上清液(FMS)后,炎症细胞因子(包括肿瘤坏死因子α (TNF-α)、IL-6、IL-8、IL-17和干扰素γ (IFN-γ))的表达显著上调,而非pSS对照组的FMS则未引起此类反应(图8A)。对这些FMS样本进行非靶向代谢组学分析显示,苯乙醛和马尿酸显著富集——这两种代谢物在pSS患者的粪便和血清中也分别高于非pSS对照组(图8B)。接下来,他们检测了四种代表性菌种:S. parasanguinis、V. parvula、L. salivarius和白色念珠菌。对这些菌种培养上清液的代谢组学分析表明,这四种菌种均能利用芳香族氨基酸及其相关底物,并且各自产生多种pSS富集代谢物。例如,S. parasanguinis和L. salivarius产生吲哚硫酸盐;L. salivarius和白色念珠菌产生胆绿素;V. parvula产生犬尿氨酸。PMBC刺激试验揭示了菌种特异性的免疫刺激谱(图8C)。L. salivarius显著上调TNF-α、IL-6、IL-8和IL-17的表达;V. parvula增加TNF-α、IL-6和IL-8的表达;白色念珠菌强烈诱导TNF-α、IL-6和IFN-γ的表达;S. parasanguinis主要升高TNF-α和IL-6的表达。这些发现表明,肠道菌群失调整体以及特定pSS富集菌群均可触发人类免疫细胞的促炎反应,为将肠道菌群改变与pSS免疫失调联系起来提供了早期机制证据。
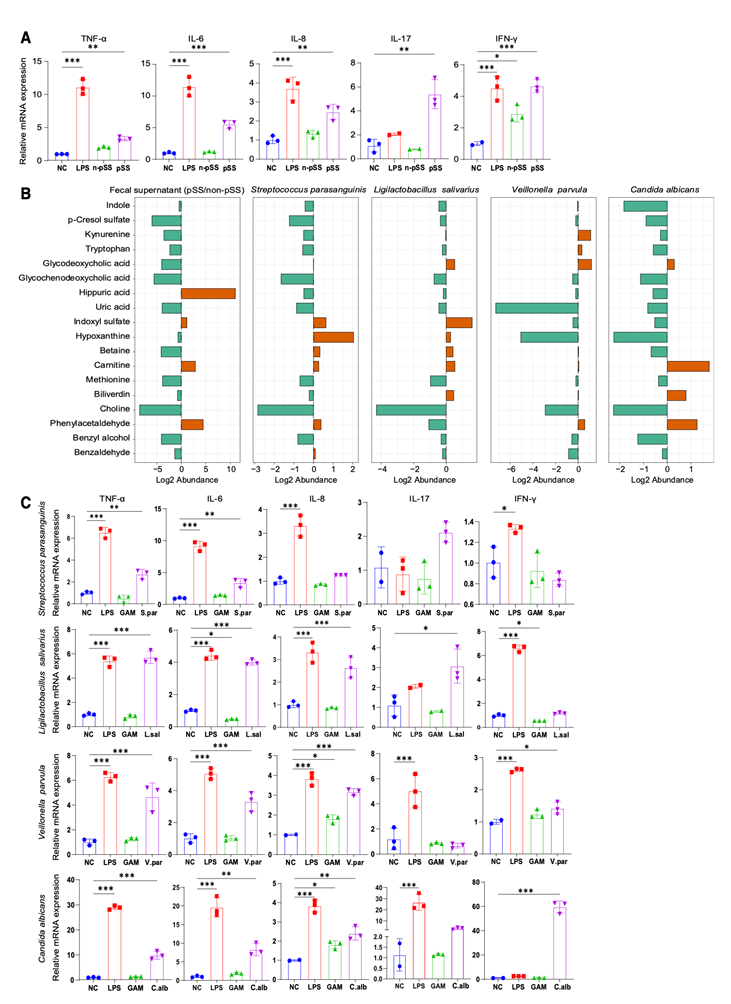
图8. 肠道菌群和原发性干燥综合征相关代表性微生物对 PBMC 的体外免疫激活作用。
(A) 用来自pSS患者和非原发性干燥综合征(非pSS)对照者的粪便微生物群上清液(FMS)刺激PBMC后,促炎细胞因子(TNF-α、IL-6、IL-8、IL-17 和 IFN-γ)的表达水平。(B) 对四种代表性pSS相关菌种的粪便微生物 (FMS) 和培养上清液进行非靶向代谢组学分析。(C) 通过 qPCR 定量分析PBMC的细胞因子反应。
09
药物对原发性干燥综合征患者肠道菌群的影响
免疫抑制剂、抗生素和生物制剂已证实可通过不同的机制调节肠道菌群,并可能影响其组成和功能。在确定pSS患者和非pSS对照组的肠道菌群组成存在显著差异后,他们进一步研究了这些差异是否受药物治疗的影响。分析显示,接受药物治疗的pSS患者(MP组)的肠道细菌多样性显著低于未接受药物治疗的pSS患者(NMP组)。虽然两组间观察到的物种丰富度没有差异,但MP组的香农指数显著降低(图9A-B),表明药物可能选择性地消耗某些对抗生素敏感的菌群,这一模式与之前的报道一致。然而,主坐标分析(PCoA)显示MP组和NMP组之间没有显著差异(图9C),表明尽管药物治疗导致多样性降低,但总体菌群组成仍然相对稳定。差异丰度分析鉴定出MP组中发生改变的特定菌群(图9D)。在MP组中,Ruminococcus sp. NSJ-71、B. dentium、Dysosmobacter welbionis和Lacrimispora celerecrescens显著富集,而Ruminococcus bromii、Phocaeicola coprophilus、Eubacterium siraeum、Adlercreutzia equolifaciens和Parasutterella excrementihominis则显著减少。这些发现表明,虽然药物不会广泛地改变肠道微生物群落结构,但它们可能对特定细菌群落施加选择压力,从而导致接受药物治疗的pSS患者肠道菌群组成发生靶向性变化。
pSS患者发生ILD的风险增加。为了确定ILD的存在是否与特定的肠道微生物特征相关,他们比较了ILD患者(pSS-ILD组)和无ILD患者(pSS-NO-ILD组)的肠道菌群。稀疏曲线显示,随着样本量的增加,pSS-ILD组的微生物丰富度略低,但两组之间的物种计数和香农多样性均无显著差异(图9E-F)。PCoA同样显示两组的整体群落组成没有差异(图9G)。尽管整体组成没有差异,但物种水平的分析发现两组之间存在一些丰度不同的分类群。在pSS-ILD患者中,Blautia wexlerae、Enterococcus casseliflavus、Bifidobacterium longum、Fusobacteriaceae bacterium和Staphylococcus epidermidis显著富集,而果胶裂毛螺菌Lachnospira pectinoschiza、Bacteroidota eggerthii和Raoultella ornithinolytica则显著减少(图9H)。这些微生物群落的变化可能反映或促进了pSS背景下间质性肺病的病理生理过程,并为未来的机制研究和生物标志物开发提供了潜在线索。
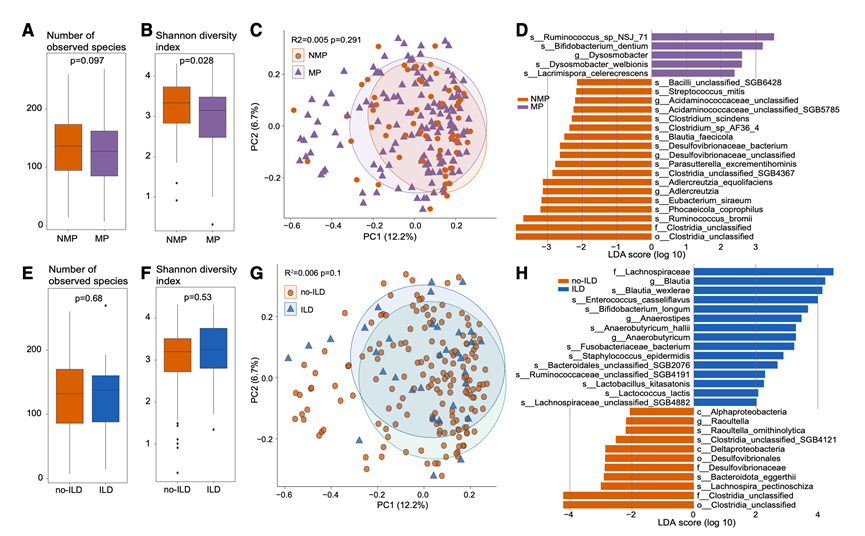
图9. 患者特征对肠道菌群多样性和组成的影响。
(A-B) 箱线图比较了接受药物治疗 (MP) 和未接受药物治疗的 pSS 患者 (MNP) 之间观察到的细菌物种丰富度和香农多样性指数。(C) 基于细菌物种水平的 Bray-Curtis 距离的 PCoA 分析,显示 MP 和 MNP 组之间存在明显的聚类差异。(D) 线性判别分析(LDA)效应量(LEfSe)条形图显示MP组和NMP组之间具有显著差异丰度的细菌物种。(E-F) 箱线图比较了患有 ILD 的 pSS 患者(pSS-ILD)和未患 ILD 的 pSS 患者(pSS-NO-ILD)之间观察到的细菌物种丰富度和香农多样性指数。(G) 基于细菌物种水平的 Bray-Curtis 距离的 PCoA 显示 pSS-ILD 组和 pSS-NO-ILD 组之间存在明显的聚类。(H) LEfSe 条形图显示 pSS-ILD 和 pSS-NO-ILD 患者之间具有显著差异丰度的细菌种类。
+ + + + + + + + + + +
结 论
本研究对 206 例 pSS 患者和 355 例非 pSS 对照组的粪便样本进行了宏基因组测序,并将组成和功能分析与血清和粪便代谢组学数据整合。pSS 与广泛的多界微生物改变相关,包括 49 种细菌、19 种真菌和 1323 种病毒。这些微生物特征形成了稳健的跨界相关性,并在独立的验证队列中实现了较高的诊断准确性。功能和代谢组学分析表明,患者体内毒素相关和芳香族代谢通路富集,而保护性代谢物减少。pSS富集细菌含有丰富的免疫原性表位、毒力因子和抗菌素耐药基因,并能在体外诱导促炎反应。这些发现共同勾勒出pSS的多方面微生物框架,并提示肠道菌群失调与免疫失调之间存在机制联系。
+ + + + +



