

 English
English文献解读|Cancer Cell(44.5):结直肠癌中KRAS抑制剂的遗传和非遗传耐药机制并存
✦ +
+
论文ID
原名:Concurrent genetic and non-genetic resistance mechanisms to KRAS inhibition in colorectal cancer
译名:结直肠癌中KRAS抑制剂的遗传和非遗传耐药机制并存
期刊:Cancer Cell
影响因子:44.5
发表时间:2026.05.21
DOI号:10.1016/j.ccell.2026.04.009
背 景
结直肠癌(CRC)是全球第二大癌症相关死亡原因,每年造成近100万人死亡。Kirsten鼠肉瘤病毒癌基因同源物(KRAS)是CRC中最常见的突变癌基因,在高达50%的病例中均可观察到。过去十年间,多种靶向RAS蛋白的选择性和非选择性小分子抑制剂相继问世。首创的KRAS抑制剂adagrasib和sotorasib已在携带KRAS G12C突变的多种癌症患者中显示出临床疗效。除了这些G12C选择性小分子外,目前还有一系列其他化合物正处于临床前和临床开发阶段,包括G12D选择性抑制剂和泛RAS抑制剂,这些抑制剂有望靶向大多数KRAS变体。因此,在接下来的十年里,很可能数百万癌症患者将接受 KRAS 抑制剂治疗。了解KRAS靶向药物原发性和获得性耐药的机制对于开发能够获得更深层、更持久疗效的联合疗法至关重要。
实验设计
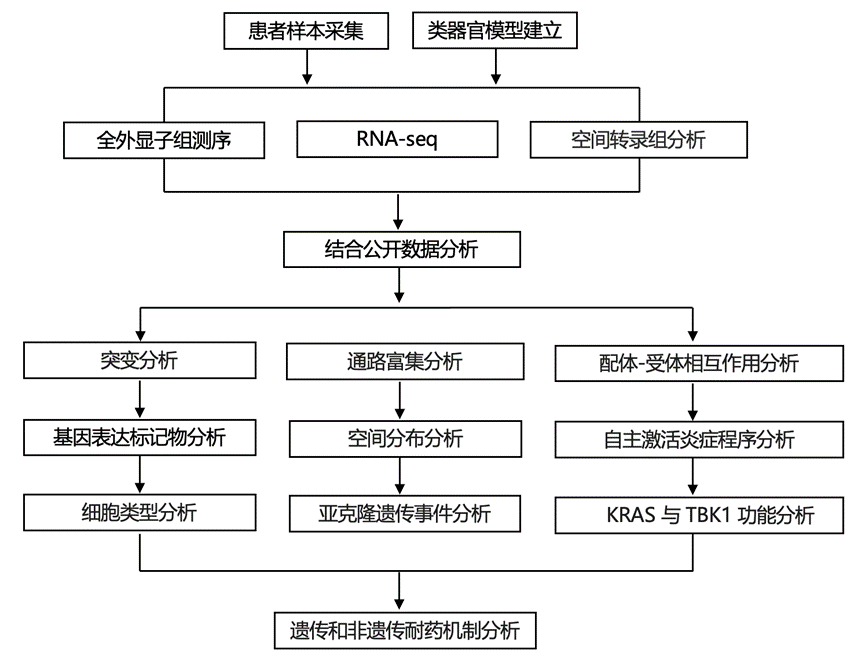
结 果
01

CRC中KRAS抑制的遗传和非遗传适应
为了确定KRAS抑制剂的遗传和非遗传反应,研究团队前瞻性地收集了12例接受KRAS G12C联合EGFR抑制剂治疗的CRC患者的治疗前(n = 11)、治疗中(第7天至第21天)(n = 11)和疾病进展后(“耐药”)(n = 8)的匹配活检样本(图1A-B)。11例患者接受了治疗前和治疗中活检,而7例患者在三个时间点接受了活检;在某些情况下,基因组分析是通过游离DNA或循环肿瘤DNA(ctDNA)而非组织活检获得的(图1A)。患者参与了两项临床试验之一,一项是索托拉西布联合帕尼单抗,另一项是阿达格拉西布联合或不联合西妥昔单抗的试验;所有患者此前均接受过氟尿嘧啶类化疗,除1例患者外,其余患者均接受了KRAS G12C和EGFR联合抑制治疗。根据RECIST标准(图1A),其中5例患者达到部分缓解,7例患者病情稳定。平均治疗持续时间为9.4个月,与更大规模的临床试验人群数据较为接近。
利用 MSK-IMPACT 进行靶向外显子组测序显示,在 11 例可获得治疗前和耐药样本(包括实体瘤或液体活检)的患者中,有 7 例存在获得性基因事件(图 1A)。其中 6 例患者在已知的 RAS 信号通路调控因子中存在一个或多个获得性突变,包括KRAS G12C和FLT3扩增,以及EGFR、FLT3、DUSP4和ROS1 的错义突变。大多数推定的耐药驱动基因是亚克隆的;在耐药活检中检测到的 17 个新发突变中,只有 7 个是克隆性的(定义为变异等位基因频率相对于预先存在的克隆突变 >50%)。其余 4 例患者未发现可识别的获得性基因事件。因此,与先前报道的 CRC 和其他癌症类型在 KRAS 抑制剂治疗进展后的情况类似,逃逸突变并非普遍存在,且通常为亚克隆突变。
为了鉴定肿瘤表型适应性,他们对匹配的治疗前、治疗中和耐药活检样本进行了空间转录组学分析(图1B)。他们采用了一种循环荧光原位杂交平台(CosMx),该平台可直接在福尔马林固定石蜡包埋(FFPE)组织上以单细胞分辨率定量950个转录本的相对丰度。所有患者样本中共有679588个细胞通过了质量控制,其中包括375985个癌细胞。对单细胞谱进行无监督聚类分析,鉴定出14个细胞亚群,并用已知的基因表达标记物进行注释(图1C-D)。根据癌细胞高表达的上皮/CRC标志物(KRT8、KRT18、KRT19、EPCAM和CLDN4)对癌细胞进行注释,这些标志物与其他转移部位的上皮细胞群(例如肝细胞)显著不同(图1D)。恶性细胞表现出显著的异质性,并通常根据患者和治疗状态进行聚类(图1E)。治疗前和耐药样本的转录组多样性高于治疗中样本,后者通常聚类在一起,且肿瘤间异质性降低(图1E)。相反,肿瘤内转录组异质性在不同治疗条件下相似(图1E)。正如预期的那样,细胞类型丰度分析显示,与治疗前相比,治疗早期恶性细胞的数量显著减少(图1F-G)。在治疗期间的活检中,癌症相关成纤维细胞 (CAF)、B 细胞和内皮细胞的比例有所增加(图1F-G),但当从分析中排除癌细胞时,只有 B 细胞的差异才具有统计学意义。
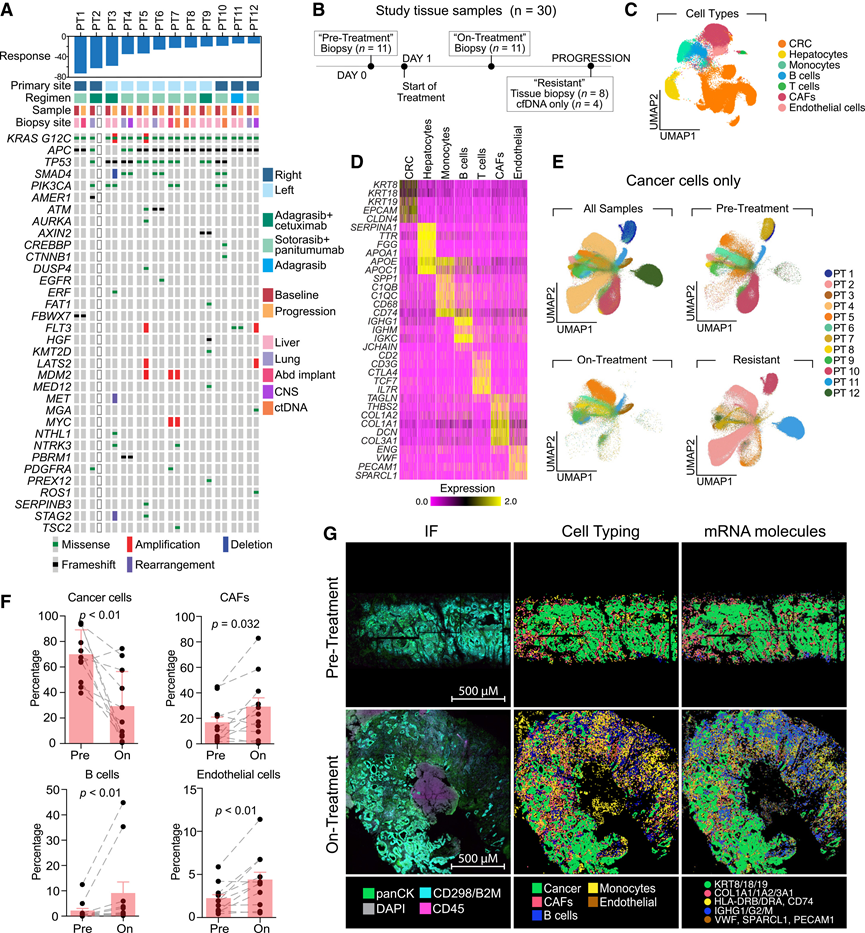
图1. KRAS和EGFR联合抑制对肿瘤微环境的影响。
(A) 接受 KRAS 和 EGFR 联合抑制治疗的 CRC 患者的肿瘤大小(RECIST 标准)、临床特征和肿瘤印迹(治疗前和进展后)的百分比变化。(B) 研究流程。(C) 按细胞类型着色的患者样本的均匀流形近似和投影(UMAP)。(D) 单细胞热图显示肿瘤区室中选定细胞类型标记的标准化表达。(E) 按治疗状态分组并按患者 ID 着色的癌细胞的 UMAP 分析。(F) 治疗前(Pre)和治疗中(On)组织活检中细胞亚群的百分比。(G) 具有代表性的治疗前和治疗中患者匹配样本。
02
癌细胞区室中 KRAS 抑制引起的转录组适应
鉴于 CosMx 平台在区分特定免疫细胞群方面的灵敏度有限,他们对部分组织量充足的样本进行了多重免疫荧光 (mIF) 分析,以补充空间转录组学分析数据。使用针对 panCK、CD4、FOXP3、CD8、CD68 和 CD163 的抗体对上皮细胞和免疫细胞亚群的相对丰度进行了定量。该分析显示,治疗期间样本中 CD4+ T 细胞显著富集,而 CosMx 检测未能有效捕获该细胞群。其他免疫细胞和基质细胞群在不同时间点之间未显示出统计学意义上的显著差异。为了鉴定肿瘤细胞对 RAS 抑制的适应性反应,他们检测了患者匹配活检样本恶性组织中差异表达的基因和程序。对 Hallmark 和经整理的肠道细胞类型特异性基因集进行过表达分析 (ORA) 和基因集变异分析 (GSVA) 显示,在治疗期间和耐药样本中,肠道潘氏细胞和干细胞特征以及多种炎症程序均发生激活(图 2A-B)。对至少三分之二样本中显著差异表达的单个基因进行注释,再次显示治疗期间样本中干扰素 (IFN) 和细胞因子信号通路显著富集(图 2C)。相比之下,耐药样本显示出上皮间质转化(EMT)、胎儿样细胞和Yes-associated protein 1 (YAP)程序的富集(图2B-D),和其他研究者此前已将这些程序与CRC转移和WNT靶向治疗耐药性联系起来。此外,KRAS抑制剂治疗还导致上皮和肠道分化标志物显著下调,以及肠道干细胞标志物相应上调,但在大多数情况下,这些标志物在耐药样本中恢复至基线水平(图2B)。正如预期的那样,与基线相比,治疗期间 KRAS 靶点的表达较低,随后在部分患者病情进展时 KRAS 靶点的表达增加(图2B-D),这可能是由于停止 KRAS/EGFR 治疗所致。
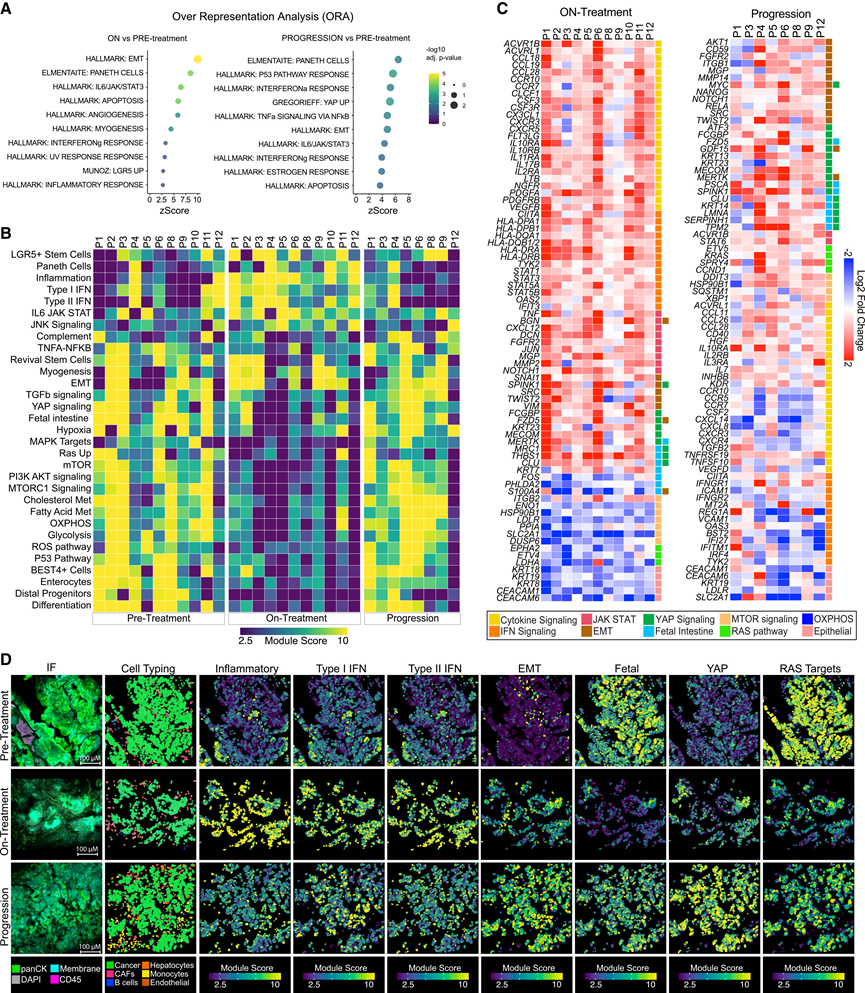
图2. 癌细胞区室中 KRAS 抑制引起的转录组适应。
(A) Hallmark 和精选的肠道细胞类型特异性基因集的 ORA 气泡图显示了治疗期间与治疗前以及耐药患者样本与治疗前样本中显著富集的十条通路。(B) 热图显示了各个患者样本中 Hallmark 和肠道细胞类型特异性基因集的模块得分分位数。(C) 治疗期间和耐药(与治疗前相比)患者匹配样本中癌细胞的差异基因表达。(D) 代表性病例(患者 6)显示免疫荧光和空间图像。
03
KRAS抑制剂的遗传性和非遗传性耐药性并存
在进展期样本中同时存在遗传和非遗传转录改变,这引发了人们对这些改变是否在单个肿瘤中同时发生的疑问。事实上,在分析的8个耐药活检样本中均发现了转录重编程,包括那些具有多个推定的遗传耐药性改变的样本(图3A-B)。例如,对患者5的耐药肿瘤进行外显子组测序显示,KRAS G12C(拷贝数增加5.9倍)、FLT3和MDM2基因扩增,以及DUSP4、ATM、AURKA、SERPINB3和STAG3基因获得性错义突变(图3A)。除了预期的KRAS转录本上调和丝裂原活化蛋白激酶 (MAPK) 信号下游重新激活(可能由KRAS G12C扩增和其他RAS 通路突变驱动)之外,转录组分析还揭示了在疾病进展过程中 YAP 和胎儿肠道特征的强烈富集(图 3 B),表明即使在具有已描述的遗传耐药谱的情况下,转录和谱系适应也可能发生。
在某些病例中,MSK-IMPACT测序和转录组学数据揭示了共存的遗传和非遗传耐药机制,但空间分析显示肿瘤内存在显著的异质性,不同适应性程序在单个肿瘤的不同区域富集(图3C-F)。例如,患者4在接受索托拉西布联合帕尼单抗治疗23.5个月后病情进展,MSK-IMPACT测序未发现任何预测可激活MAPK信号通路的遗传事件(图3C)。空间转录组学分析揭示了两个具有看似互斥的转录谱的独特区域。区域1显示I型干扰素、YAP和胎儿肠道程序显著富集,但MAPK信号通路活性较低。相反,区域2显示MAPK信号通路显著激活,但I型干扰素、YAP和胎儿标志物受到抑制(图3D-E)。他们在患者 1 的进展期活检中观察到了类似的现象,表现为孤立的“YAP 高表达、MAPK 低表达”肿瘤区域与“YAP 低表达、MAPK 高表达”区域相邻。这些观察结果凸显了同一肿瘤内可能平行演化出独立的耐药谱,并可能解释了通过 cfDNA 分析检测到的进展期多个亚克隆遗传事件的频繁出现。
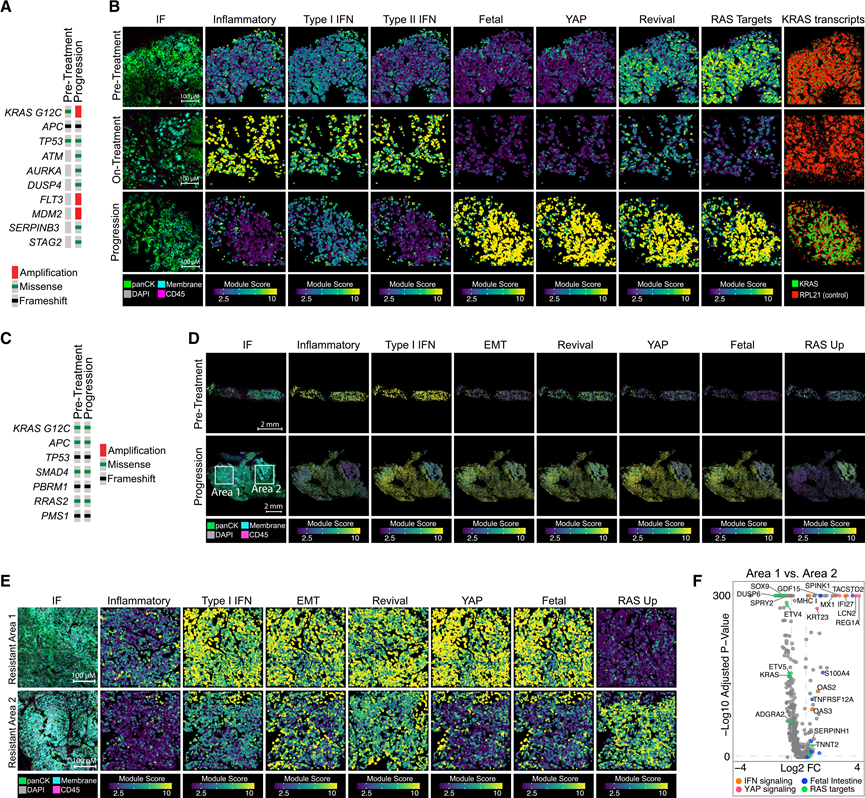
图3. KRAS抑制的共存遗传和非遗传耐药机制。
(A) 患者 5 治疗前和进展后活检的肿瘤印迹图。(B) 患者 5 治疗前和耐药活检的免疫荧光和空间图像。(C) 患者 4 治疗前和进展后活检的肿瘤印迹图。(D) 低倍镜下观察到的4号患者的免疫荧光和空间图像。(E) 图 (D) 中的区域 1 和区域 2 以更高的放大倍数显示。(F) 火山图显示区域 1 与区域 2 中差异表达的基因。
04
KRAS抑制后癌细胞自主激活炎症程序
接下来,他们探究了转录重编程是否依赖于肿瘤微环境。对非恶性细胞区室的通路和差异基因表达分析显示,在治疗期间和耐药样本中,CAF、髓系细胞和淋巴系细胞均表现出广泛的炎症特征(图S6,图S7)。过表达分析表明,细胞因子、IFN、JAK-STAT和免疫相关通路在多个细胞区室中显著富集。值得注意的是,CXCL12(一种先前与EMT和治疗耐药性相关的趋化因子)是治疗期间和耐药时间点CAF和单核细胞中上调最显著的基因之一(图S6,图S7)。
为了研究潜在的细胞间通讯,他们使用 CellChat 进行了配体-受体相互作用分析。CellChat 通过估算“发送细胞”中配体和“接收细胞”中相应受体的表达水平来推断细胞间信号传导。为了深入了解配体-受体相互作用的时间和状态特异性,对治疗前、治疗中和疾病进展期的样本,以及 IFN 高表达、YAP 高表达和 RAS 高表达的活检区域进行了独立的 CellChat 分析(图 S8)。这些分析揭示了一些具有统计学意义但强度较弱的配体-受体相互作用,包括髓系细胞和淋巴细胞到癌细胞的相互作用,例如在治疗期间,T 细胞到肿瘤细胞的潜在 TGFB2-TGFBR2 信号传导(图 S8)。有趣的是,之前已发现TGF-β信号通路是肠癌细胞谱系改变和耐药性的驱动因素,这表明此类相互作用可能在此背景下具有重要的功能意义。然而,总体而言,这些数据表明免疫细胞到癌细胞的直接配体-受体信号传导可能受到限制。当然,CellChat分析仅限于包含在950个基因的CosMx panel中的配体-受体对;不能排除未包含在该panel中的基因/蛋白质参与信号传导的可能性。
为了在完整肿瘤微环境存在或不存在的情况下直接测量肿瘤细胞对KRAS抑制剂的反应,他们构建了携带Apc截断突变(APC Q1405X)、内源性Kras突变(KRAS G12C)和Trp53截断突变(p53 Q97X)(以下简称AKP-G12C)的C57Bl/6结肠类器官。将类器官移植到免疫功能正常(C57Bl / 6)和免疫缺陷(NSG)小鼠体内后,均显示出对adagrasib治疗的显著反应,并且在4周内,药物存在下类器官的生长速度加快。他们观察到C57Bl/6小鼠和NSG小鼠的肿瘤生长没有显著差异,这表明至少在小鼠模型中,adagrasib的反应和耐药性并不依赖于免疫细胞类型的信号传导,不能排除非免疫细胞群的作用。为了在排除所有其他细胞类型的情况下探究耐药性,他们用adagrasib单独处理AKP-G12C类器官,或用adagrasib联合afatinib处理AKP-G12C类器官,模拟临床上使用的KRAS/EGFR抑制剂联合治疗。AKP-G12C类器官培养物显示ERK磷酸化水平呈剂量依赖性降低(图4 A),并且在KRAS抑制(adagrasib)或KRAS/EGFR联合抑制(adagrasib/afatinib)后出现生长停滞(图4B-D)。持续治疗10-12周后,产生了高度耐药的类器官(图4B-F),与未经治疗的类器官相比,EC50值提高了25至100倍(图4C-D)。Adagrasib耐药的类器官对KRAS G12C抑制剂sotorasib以及近期报道的多选择性RAS(ON)抑制剂RMC-7977也表现出交叉耐药性。
为了表征KRAS抑制的反应,他们对AKP-G12C类器官在治疗前、治疗期间[单独使用adagrasib (100 nM) 或adagrasib/afatinib (50/5 nM) 治疗72小时]以及耐药细胞(模拟患者活检的情况)进行了RNA-seq。总共构建了5个独立的adagrasib耐药和5个adagrasib/afatinib联合耐药的AKP-G12C细胞系。部分耐药类器官细胞系表现出高拷贝数Kras扩增(定义为≥4个拷贝)(图4G)。有趣的是,Kras扩增的类器官对KRAS抑制具有依赖性,因为停药会导致类器官生长减少。这些发现与KRAS G12C扩增的人类CRC在停药后观察到的衰老表型相似。对未经药物治疗的阿达格拉西布和阿达格拉西布/阿法替尼处理的类器官进行转录组分析,结果显示肠道干细胞特征富集,以及炎症程序(包括I型和II型干扰素)的富集,这些炎症程序在耐药类器官中减少(图4H-I)。这些发现与患者活检数据相符,表明KRAS抑制后炎症程序早期激活,并且并非严格依赖于肿瘤微环境中的非恶性细胞。YAP和胎儿样特征在耐药类器官的转录谱中占主导地位,且与KRAS拷贝数改变无关,这表明遗传和非遗传改变共存,与临床病例中观察到的情况一致(图4H-K)。事实上,与患者样本相比,治疗中和耐药的AKP类器官中均观察到基因集富集存在显著的正相关性。与YAP转录输出的增加一致,耐药类器官中活性(非磷酸化)YAP的水平高于亲代类器官(图4 J);急性阿达格拉西布/阿法替尼治疗后,未观察到非磷酸化YAP水平升高(图4 J)。
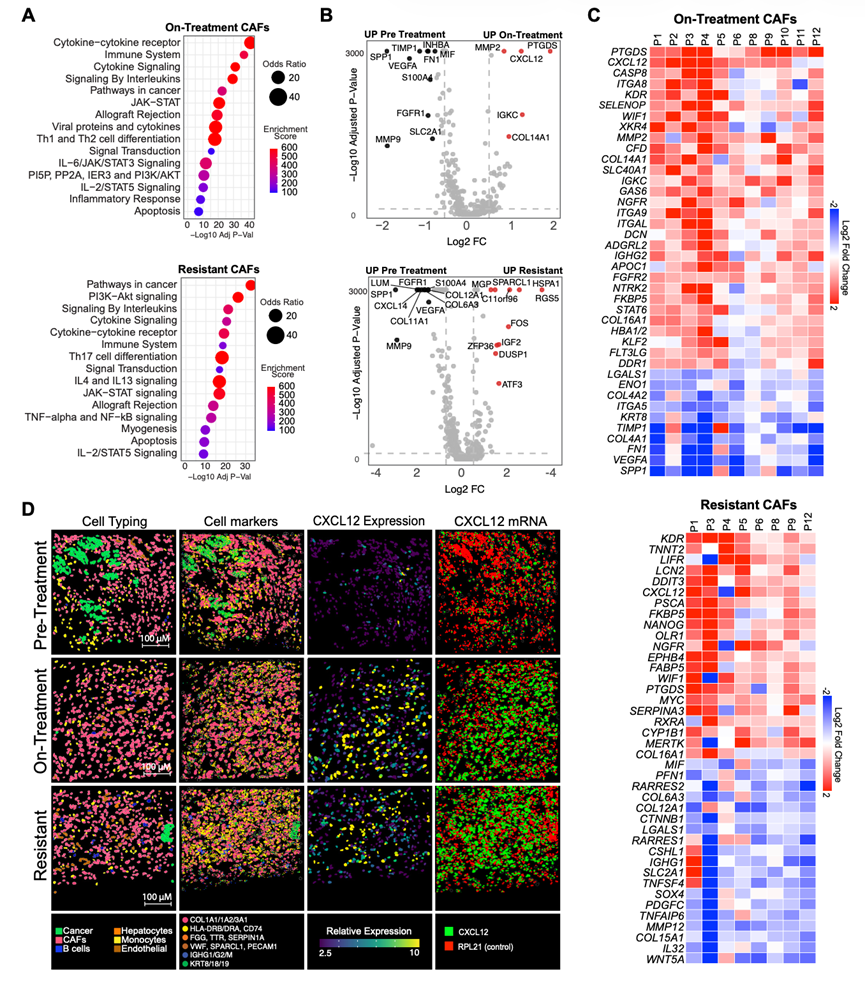
Fig. S6 癌症相关成纤维细胞对KRAS抑制的转录组适应。
(A) ORA气泡图展示了在治疗前后以及耐药与非耐药患者样本中,来自Hallmark、Reactome和KEGG基因集集合的五个显著富集通路。(B) 火山图显示治疗前后样本中与治疗前时间点相比差异表达的基因。(C) 热图显示治疗前后CAF细胞中差异表达基因的变化。(D) 代表性病例(患者9)在不同治疗时间点显示CAF中CXCL12的相对和绝对表达情况。
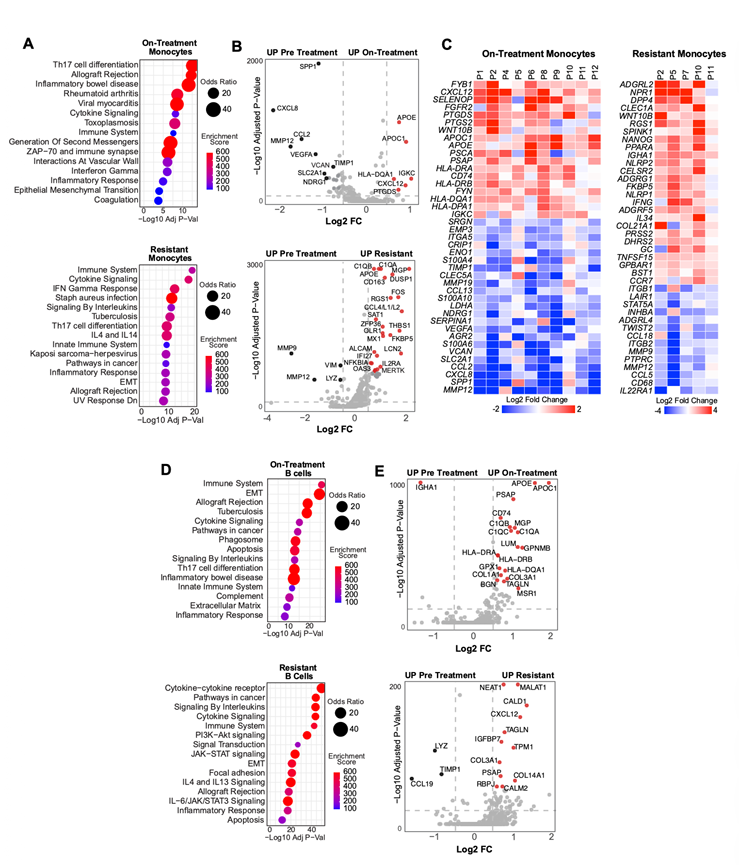
图S7. 髓细胞和B细胞对KRAS抑制的转录组适应。
(A) ORA气泡图展示了在治疗前后以及耐药与非耐药髓系细胞中,来自Hallmark、Reactome和KEGG基因集的五个显著富集通路。(B) 火山图显示髓系细胞中治疗前后不同时间点的治疗组与耐药组基因表达差异。(C) 显示治疗组与耐药单核细胞中差异表达基因的热图。(D) ORA气泡图展示了在B细胞治疗前后以及耐药与非耐药状态中,来自Hallmark、Reactome和KEGG基因集集合的五个显著富集通路。(E) 火山图显示B细胞在治疗前后不同时间点的治疗组与耐药组中差异表达基因。
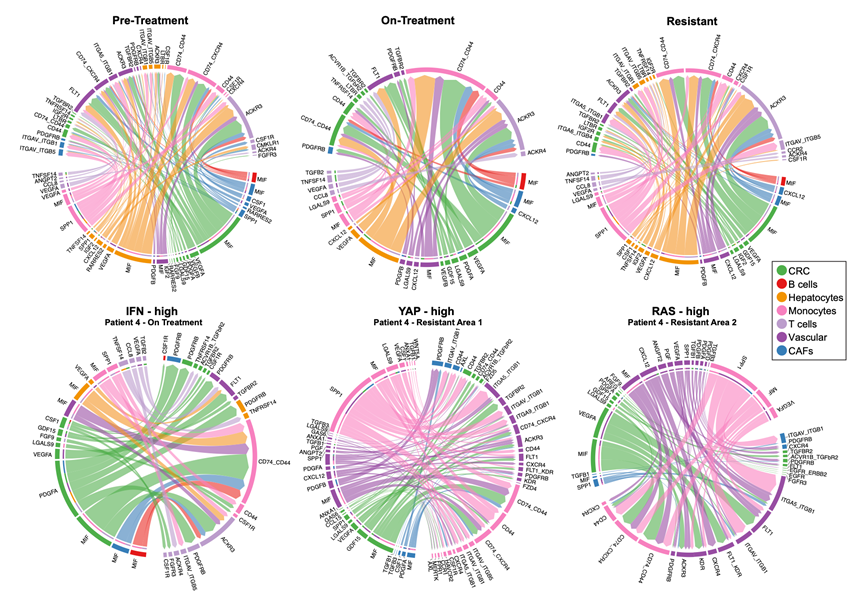
图S8. CellChat 和弦图显示配体-受体相互作用。
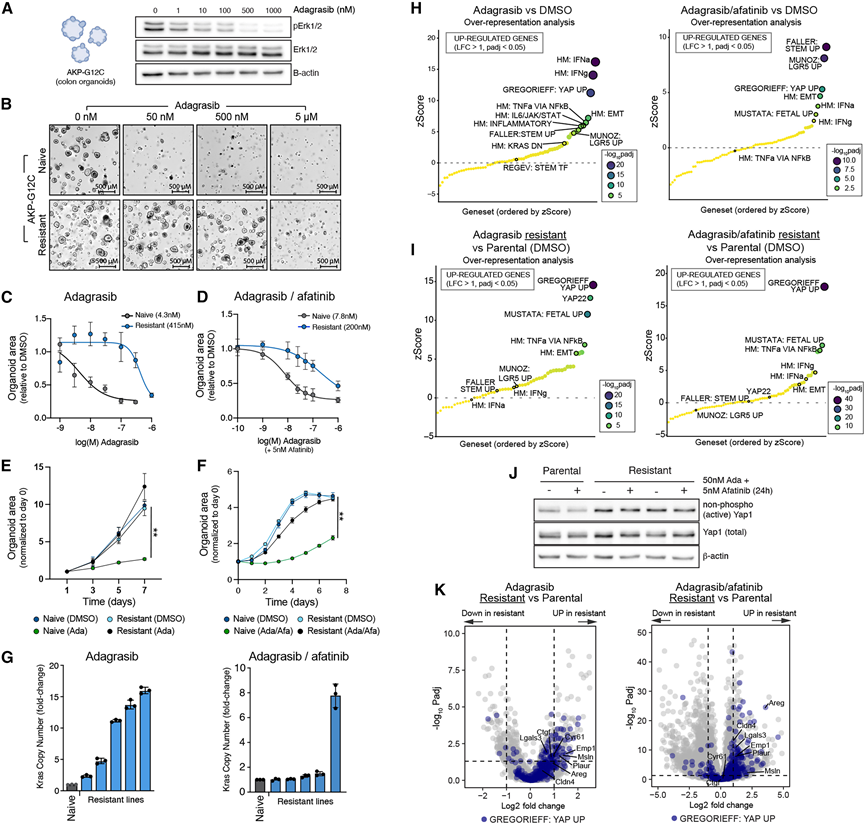
图4. 转录组对 KRAS 抑制的适应是癌细胞自主的。
(A) 用adagrasib(0-1,000 nM)处理24小时后,亲代AKP-G12C类器官中活性和总ERK的Western blot分析。(B) 用递增浓度的阿达格拉西布处理亲代和KRAS抑制剂耐药的AKP-G12C类器官的代表性明场图像。(C) 用递增浓度的阿达格拉西布处理亲代和阿达格拉西布抗性AKP-G12C类器官,观察其生长情况。(D) 用递增浓度的阿达格拉西布和阿法替尼处理亲代和阿达格拉西布/阿法替尼耐药的AKP-G12C类器官的生长情况。(E) 用阿达格拉西布的 EC80(100 nM)处理亲代和阿达格拉西布抗性 AKP-G12C 类器官(KRAS 拷贝数低)的生长情况。(F) 用 adagrasib 和 afatinib 的 EC80(adagrasib 50 nM 和 afatinib 5 nM)处理亲代和 adagrasib/adafatinib 耐药的 AKP-G12C 类器官的生长情况。(G) 亲代和 KRAS 抑制剂抗性 AKP-G12C 类器官的 KRAS 拷贝数。(H) 过表达分析(ORA)气泡图显示,在用 KRAS 抑制剂(adagrasib 100 nM)或 KRAS 联合 EGFR 抑制剂(adagrasib 50 nM,afatinib 5 nM)处理的亲代 AKP 类器官中,Hallmark 基因集和精选的肠道基因集均富集。(I) ORA气泡图显示抗性AKP类器官与亲本AKP类器官中基因集的富集情况。(J) 用 DMSO 或 adagrasib 加 afatinib 处理 24 小时后,对亲代和耐药的 AKP-G12C 类器官进行非磷酸化和总 Yap1 的免疫印迹分析。(K) 火山图显示了抗性 AKP-G12C 类器官与亲本类器官中差异表达基因的分布。
05
TBK1抑制剂可阻断早期炎症重编程,并与KRAS抑制剂产生协同作用
在癌细胞对靶向治疗和细胞毒性疗法的初始反应中,炎症反应激活。为了检验阻断炎症信号是否能消除初始适应性反应,并与KRAS抑制剂协同控制肿瘤细胞生长,他们对患者来源的KRAS G12C突变型结直肠癌类器官(患者来源类器官或PDO)进行了一项小规模、针对性的药物筛选,将阿达格拉西布与靶向炎症通路的激酶抑制剂联合使用。他们评估了两种浓度(0.5和1 μM)的小分子药物对八种激酶(PI3K、FAK、KIT、JNK、IKK、TGFβRI、JAK和TBK1)的靶向作用(图5A)。在这些抑制剂中,只有TANK结合激酶1 (TBK1) 抑制剂TBK1/IKKε-in-5在adagrasib存在的情况下延缓了类器官的生长(图5A),而单独抑制TBK1对类器官的生长没有显著影响。FDA批准的双重TBK1/JAK抑制剂momelotinib也观察到了类似的结果(图5B)。与TBK1在细胞对KRAS抑制的反应中发挥作用相一致,用adagrasib处理AKP-G12C类器官24-72小时后,TBK1磷酸化水平呈剂量依赖性增加(图5C)。在用多选择性RAS抑制剂RMC-7977处理的人类KRAS G13D突变型HCT116 CRC细胞中也观察到了同样的剂量依赖性pTBK1增加。
TBK1及其相关激酶IKKε通过干扰素调节因子3 (IRF3)传递信号,IRF3是先天性抗病毒免疫反应的关键介质。事实上,在RMC-7977处理的HCT116细胞中,IRF3磷酸化水平升高,同时pTBK1水平也升高。对患者匹配样本的分析显示,CosMx panel中包含的IRF3靶基因在KRAS和EGFR联合抑制后表达上调(图5 D)。类似地,用adagrasib处理的AKP-G12C类器官的差异基因表达分析显示IRF3靶基因显著富集(图5 E),而与momelotinib联合治疗则显示炎症特征显著下调,其中IRF3靶基因的抑制最为显著(图5 F-G)。这些数据共同支持了TBK1-IRF3信号通路在KRAS抑制后激活的观点。为了确保adagrasib和TBK1抑制剂的联合作用并非仅限于单一的PDO细胞系,分别用携带KRAS G12C或KRAS G12D突变的AKP小鼠类器官、另一种KRAS G12C突变的CRC PDO(图S10 G)以及HCT116 CRC细胞进行治疗。在所有情况下,TBK1阻断作为单药治疗效果甚微,但与KRAS抑制剂联合使用时可抑制细胞增殖。TBK1/IKKε-in-5和momelotinib除了抑制TBK1外,还抑制相关的激酶;然而,仅使用两种独立的强力霉素诱导型短发夹RNA(shRNA)沉默TBK1即可模拟药物治疗的效果(图5H-I),这表明TBK1抑制足以增强阿达格拉西布对未经药物处理的类器官的活性。重要的是,momelotinib也改善了目前CRC标准联合疗法KRAS/EGFR抑制剂的疗效(图5J-K)。总之,这些数据强调,KRAS抑制后炎症程序的急性诱导,至少部分是通过TBK1介导的,可能在耐药性肿瘤的出现中发挥作用。
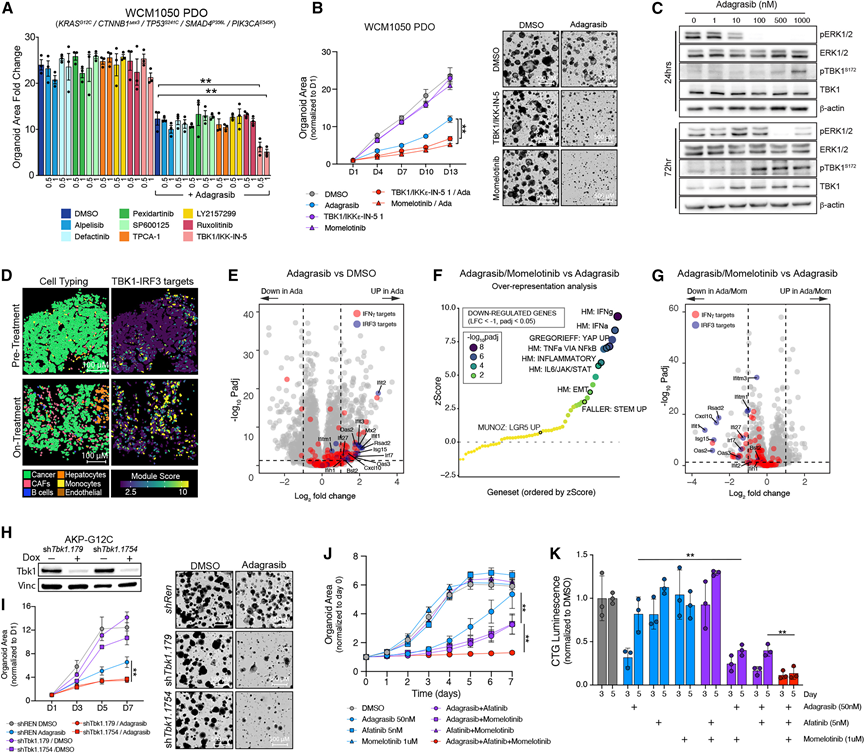
图5. 通过阻断TBK1靶向早期炎症重编程与KRAS抑制具有协同作用。
(A) 柱状图显示了PM1050 PDOs分别用阿达格拉西布(50 nM)、靶向炎症程序的激酶抑制剂或二者联合处理后的药物筛选结果。(B) 用 KRASi(adagrasib 50 nM)、TBK1i(TBK1/IKK-in-5 1 μM,momelotinib 1 μM)或二者组合处理 PM1050 类器官的生长曲线和明场图像。(C) 用adagrasib(0-1,000 nM)处理亲代AKP-G12C类器官24和72小时后,检测活性和总ERK和TBK1的免疫印迹。(D) 代表性病例(患者 5)显示治疗前和治疗中样本的图像。(E) 火山图显示了用adagrasib(100 nM)处理与用DMSO处理的亲代AKP-G12C类器官中差异表达的基因。(F) 过表达分析(ORA)气泡图显示,与单独使用adagrasib相比,用adagrasib(100 nM)加momelotinib(500 nM)处理的亲代AKP类器官中Hallmark和精选肠道基因集的富集情况。(G) 火山图显示了用adagrasib(100 nM)和momelotinib(500 nM)处理的亲代AKP-G12C类器官与单独用adagrasib处理的类器官相比,差异表达基因的分布情况。(H) 用靶向 Tbk1 的强力霉素诱导 shRNA(shTbk1)转导的 AKP-G12C 类器官的免疫印迹。(I) 用阿达格拉西布(100 nM)、强力霉素(1 μg/mL)或二者联合处理 shTbk1 和 shRen(对照)AKP-G12C 类器官的生长曲线和明场图像。(J) 用阿达格拉西布、阿法替尼、莫美替尼或组合治疗的 AKP-G12C 类器官的生长曲线。(K) 用阿达格拉西布、阿法替尼、莫美替尼或三者联合处理AKP-G12C类器官后,第3天和第5天的Cell Titre Glo (CTG)发光值。
+ + + + + + + + + + +
结 论
本研究对接受KRAS G12C和EGFR抑制剂联合治疗的患者匹配的CRC活检组织进行了靶向外显子组测序和空间转录组学分析。结果表明,大多数患者在疾病进展期均可检测到获得性基因事件,但这些事件通常是亚克隆性的,并且与转录适应状态共存。间质、YAP和胎儿样转录特征在耐药肿瘤中占主导地位,而炎症程序在治疗早期诱导。单细胞空间分析揭示了肿瘤内异质性,单个肿瘤的不同区域存在多种适应状态。利用人和小鼠类器官模型,本研究发现药物诱导的炎症程序至少部分是癌细胞自主的,并且先于耐药性的出现,确定 TBK1 为靶点,可消除炎症适应期并增强对 KRAS 抑制的反应。
+ + + + +



