

 English
English文献解读|Cell Discov(13.0):蛋白质组学对泛黑色素瘤生物学和治疗的见解
✦ +
+
论文ID
原名:Proteogenomic insights into the biology and treatment of pan-melanoma
译名:蛋白质组学对泛黑色素瘤生物学和治疗的见解
期刊:Cell Discovery
影响因子:13.0
发表时间:2024.07.23
DOI号:10.1038/s41421-024-00688-7
背 景
黑色素瘤是最常见的皮肤癌之一,转移率高,预后差。黑色素瘤起源于位于表皮基底层的色素细胞,黑色素瘤在皮肤上很常见,可根据肿瘤位置分为无毛发皮肤(手掌、脚底)(肢端黑色素瘤:AM)或非肢体(皮肤黑色素瘤:CM)。黏膜黑色素瘤 (MM) 占西方人群黑色素瘤的 0.8%–3.7% ,占中国人群黑色素瘤的 20%–30%。MM 在发现时已是晚期,与 CM 相比,治疗难度更大,因此了解其分子发病机制对于改善其诊断和治疗至关重要。
实验设计
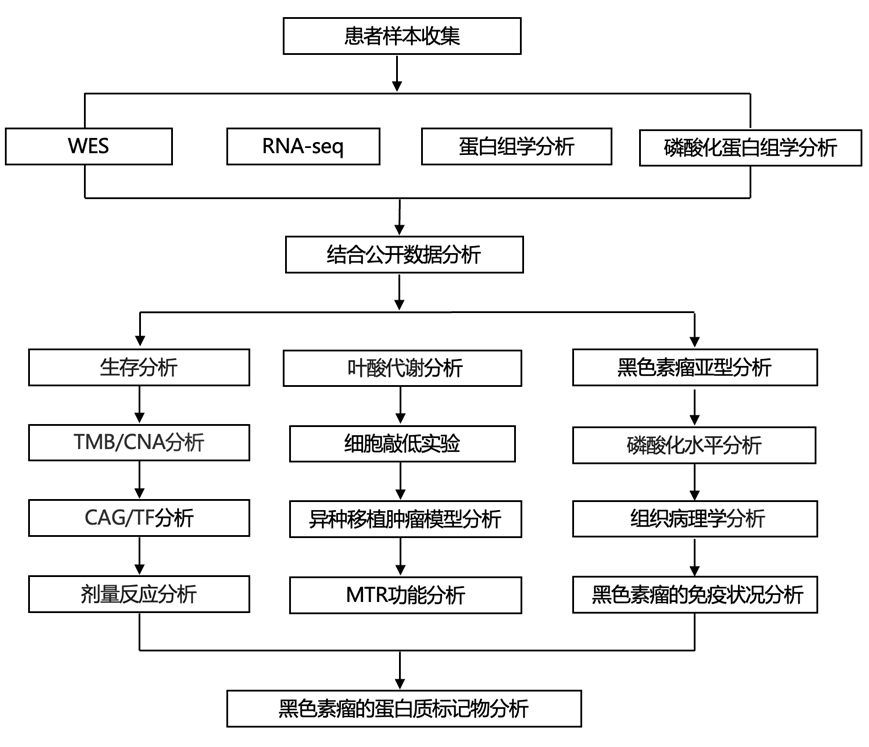
结 果
01

黑色素瘤的蛋白质组学概况
研究团队收集了155 例黑色素瘤患者的组织,其中包括 137 例原发性黑色素瘤 (PM)(81 例 AM;28 例 CM,28 例 MM)和 27 例转移性黑色素瘤,并将 43 例痣组织纳入本研究的队列作为良性对照(图1a-b)。他们对这些样本进行了全外显子组测序 (WES)、转录组学、蛋白质组学和磷酸化蛋白质组学分析(图1c)。单核苷酸变异(SNV)的总体比例与 TCGA 队列中观察到的相似,其中胞嘧啶到胸腺嘧啶 (C > T) 转换是最常见的 SNV(图1d)。基于来自TCGA队列、澳大利亚队列和其他公开数据的比较分析证实,在AM、CM和MM中,热点基因的突变频率不同(图1e)。例如,BRAF突变频率在CM中超过50%,在MM中低于5%。
他们在黑色素瘤患者中确定了四个突变特征,SBS30、SBS1、SBS18 和 SBS7a,与患者诊断时的年龄、紫外线损伤和 DNA 损伤修复有关(图1f)。根据富集分数将患者分为 SBS 阳性 (SBS+) 和 SBS 阴性 (SBS–) 组。生存分析显示,只有 SBS7a 与总生存率 (OS)相关(图1g)。在 TCGA 队列中,SBS7a 富集分数升高的患者预后较差(图1g)。DNA修复通路在SBS7a+组中显著富集,并且PRKDC、POLD4、POLK和ATR等蛋白质在SBS7a+组中显著过度表达(图1h-i)。与此一致的是,SBS7a+ 组的样本显示出明显更高的肿瘤突变负荷 (TMB)(图1j)。有趣的是,拷贝数改变(CNA)和多组学数据的相关性分析表明,SBS7a+组的样本比SBS7a– 组的样本有更多的顺式效应事件,凸显了CNA在SBS7a+组中的重要作用(图1k)。
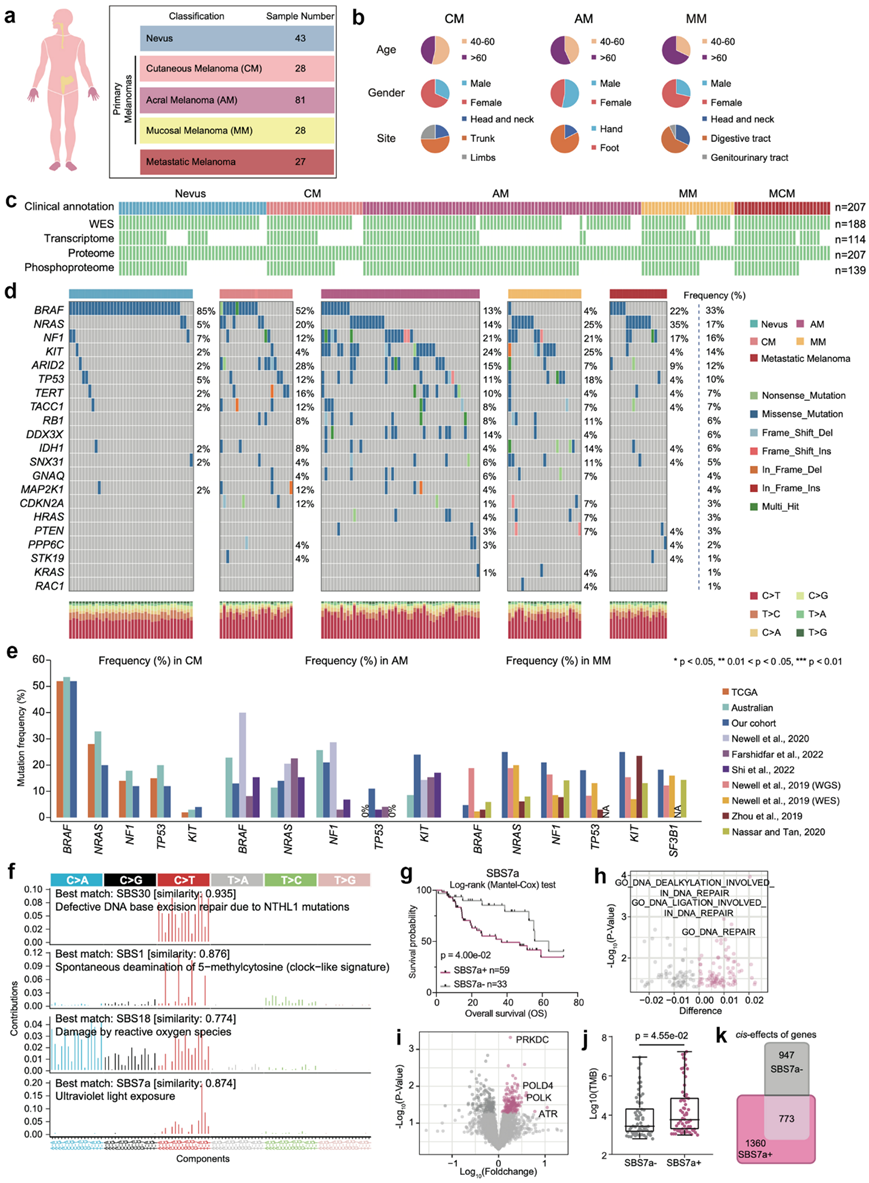
图1. 黑色素瘤样本的多组学景观。
(a) 本研究中生成的数据摘要 (b) 本研究中描述的黑色素瘤患者样本的关键人口统计学和组织学特征的饼图。(c) 黑色素瘤多组学分析示意图。 (d) 队列中黑色素瘤热点突变图。(e) 突变频率分析。(f) 相对突变频率分析。(g) 总生存率 (OS) 的 Kaplan-Meier 曲线。(h-i) 火山图显示具有 SBS7a 突变特征的患者的蛋白质通路升高。(j) 肿瘤突变负担 (TMB)分析。(k) 具有和不具有 SBS7a 突变特征的患者中 CNA变化。
02
综合蛋白质组学分析揭示突变和CNA的功能后果
基因的CNA与临床结果密切相关,为了在本研究的黑色素瘤队列中确定CNA区域中功能重要的基因,他们聚焦于663个癌症相关基因(CAG),共观察到163个RNA和蛋白质显著正相关,18个CAG[编码13个激酶(PRKACB、TAOK3、PRKDC等)和3个转录因子(TF)(PSIP1、TNFAIP3和FOXO3)]显示与患者生存强相关(图2a-c)。在这18个CAG中,MTSS1和PRKDC是与患者预后不良显著相关的关键基因,PRKDC编码激酶DNA依赖性蛋白激酶催化亚基(DNA-PKcs),其与本研究队列和TCGA队列中同源RNA和蛋白质的表达呈显著正相关(图2d)。进一步的生存分析表明,具有PRKDC扩增的患者预后较差(图2e)。他们进一步对本队列的基线数据进行了多变量Cox回归分析,包括年龄、性别、临床变量,如组织学类型、病理亚型、肿瘤部位、Clark等级、溃疡。结果显示,PRKDC扩增是黑色素瘤患者预后最重要的预测因素(图2f)。
然后,他们重点研究了PRKDC扩增对其同源蛋白和其他蛋白的功能影响。综合分析显示,除了上调其同源 mRNA 和蛋白的表达外,PRKDC扩增还增加了参与 DNA 修复、DNA 错配修复和细胞对 DNA 损伤刺激反应的蛋白质表达(图2g)。PRKDC的蛋白表达与非同源连接DNA修复过程的GSVA评分显著相关,参与DNA损伤修复的蛋白的表达,包括ATM、ATR、CUL4B、XRCC4、XRCC1和XPA,也与PRKDC的蛋白表达高度相关(图2h-i)。生存分析表明,12种DNA损伤相关蛋白,包括2种激酶(ATM和ATR),与较差的生存率相关(图2i)。在PRKDC扩增的患者中,ATM、ATR 和 PRKDC 的 mRNA 表达和推断的激酶活性显著上调,表明 PRKDC 可能与 ATM 和 ATR 发生协同效应,影响 DNA 损伤修复过程并导致不良预后(图2j-k)。
值得注意的是,由于 PRKDC 也是 SBS7a+组中表达最显著的蛋白质之一,他们评估了针对 SBS7a +患者的 PRKDC 的临床相关性。因此,他们从 SBS7a +和 SBS7a-患者收集了原代肿瘤细胞培养物 (PDC),并评估了 PDC 对 PRKDC 抑制剂 (NU7441) 的反应。结果显示,PRKDC 抑制剂显著降低了SBS7a+患者来源的 PDC 增殖,但对SBS7a-患者来源的 PDC 生长没有显著影响(图2l)。
与此结果一致,他们观察到SBS7a+患者的PDC对PRKDC抑制剂也更敏感,IC50值显著降低(图2m)。目前,化疗药物5-氟尿嘧啶(5-FU)的单独使用疗效不佳,且副作用大。他们接着研究了PRKDC抑制剂能否作为5-FU的补充剂用于SBS7a+患者的治疗。他们比较了单独用5-FU治疗和同时用5-FU和PRKDC抑制剂治疗对PDC细胞增殖率的影响。结果显示,SBS7a +患者的PDC在用PRKDC抑制剂和5-FU联合治疗后增殖率显著低于单独用5-FU治疗。然而,对于 SBS7a -患者的 PDC,与单独使用 5-FU 治疗相比,联合治疗策略并未提高细胞生长抑制效率(图2n)。这些结果表明 SBS7a+ 患者可能受益于 PRKDC 抑制剂和 5-FU 的联合治疗。
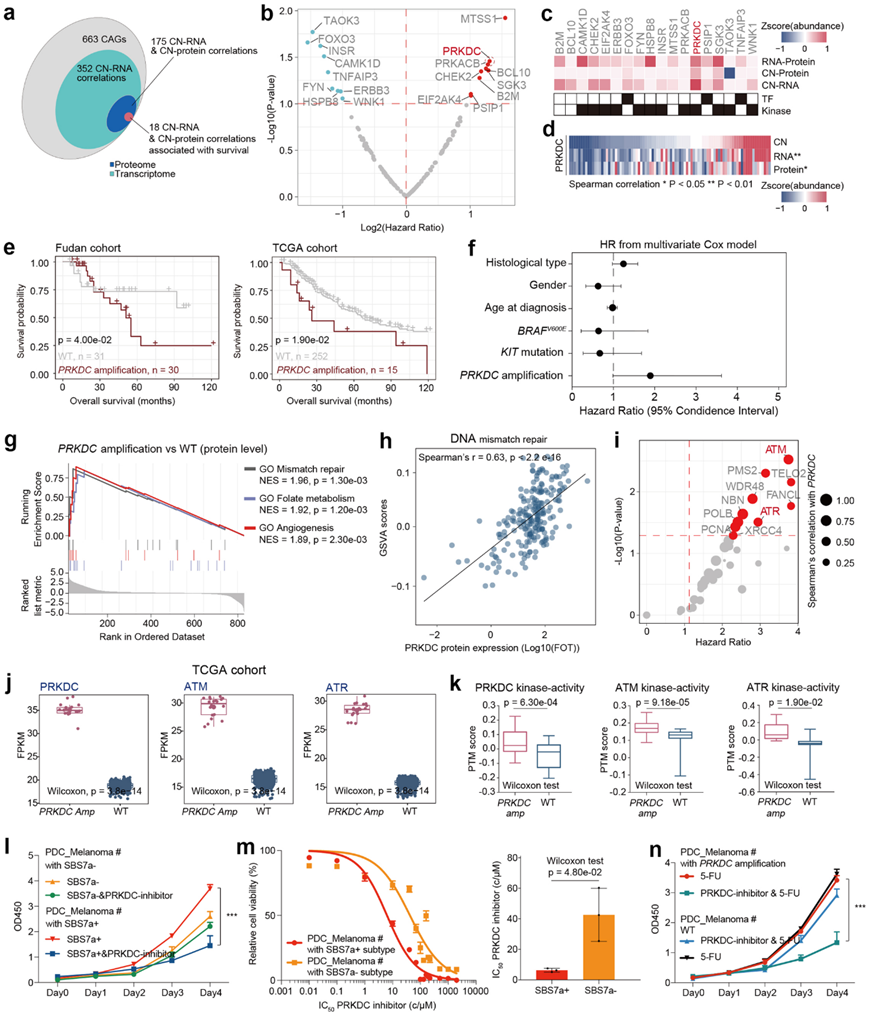
图2. 黑色素瘤样本的综合组学分析。
(a) 维恩图描绘了黑色素瘤中癌症相关基因 (CAG) 的 CNA 的级联效应。(b) 火山图显示了预测黑色素瘤 OS 的拷贝数变异。(c) 热图表示 18 种与生存相关的 CAG,这些 CAG 与拷贝数 (CN)、mRNA 或蛋白质显著相关。(d) 热图显示了 PRKDC 的 CN、mRNA 和蛋白质之间的相关性。(e) 生存曲线分析。(f) 森林图显示了基于本研究队列的PRKDC扩增和其他协变量的 Cox 回归风险比系数的 95% 可信区间。(g) PRKDC扩增与 WT 比较中 DNA 错配修复相关通路的 GSEA 图。(h) PRKDC蛋白表达的 Spearman 相关性和 DNA 修复过程的 GSVA 评分。(i) 火山图显示蛋白质的表达与PRKDC拷贝数相关并可预测黑色素瘤的 OS。(j) TCGA队列中携带PRKDC扩增子和 WT 样本的患者之间 ATM 和 PRKDC 的 mRNA 表达比较。(k) 激酶活性比较。(l)剂量反应曲线。(m) 细胞活力分析。(n) PDC 增殖情况分析。
03
叶酸代谢平衡异常导致原发性黑色素瘤肿瘤发展
PRKDC 是一种重要的 DNA-PK 。为了鉴定 PRKDC 的预后相关底物,他们对与 PRKDC 表达正相关的磷酸化底物进行了生存分析。他们筛选出了MXD3 的 S57 磷酸化位点,它是与 OS 相关的 PRKDC 中排序最高的磷酸化底物(图3a)。作为 MXD 家族的成员,TF MXD3 在细胞周期进程和细胞增殖中起着关键作用。然后,他们使用 GSVA 算法根据其靶基因 (TG) 的 mRNA 表达推断 MXD3 TF 活性。正如预期的那样,推断的 MXD3 TF 活性与 MXD3/S57 的丰度密切相关(图3b)。为了深入了解 MXD3 的 TF 活性如何导致不良预后的机制,他们进行了相关性分析,并观察到 TYMS 和 MTHFD2 是 MXD3 的关键 TG,其 mRNA 表达与 MXD3 的 TF 活性及其同源蛋白的表达密切相关(图3c)。一致地,在存在PRKDC扩增的样本中,MTHFD2 和 TYMS 的蛋白质表达显示出升高(图3d)。
功能上,TYMS 和 MTHFD2 均参与叶酸循环,叶酸循环是叶酸代谢的重要分支,可为嘌呤和胸苷的合成提供一碳单元。为了进一步说明叶酸循环对肿瘤发展的影响,他们筛选了参与叶酸代谢的酶的表达。除 MTHFD2 和 TYMS 外,SHMT2(参与丝氨酸分解代谢的关键线粒体酶,将丝氨酸转化为甘氨酸和一碳单元在PRKDC扩增患者中表现出表达显著增加(图3d)。在叶酸代谢中,MTR作为叶酸和蛋氨酸循环的独特代谢连接物,其表达在PRKDC扩增的患者中显著降低。生存分析表明MTHFD2和TYMS与预后不良相关(图3e)。虽然MTR与生存率没有表现出很强的相关性,但它与MTHFD2和TYMS结合起来发挥了很强的协同作用。MTHFD2/TYMS表达在MTR低表达组中的生存率低于在MTR高表达组中的生存率(图3f)。
他们利用CCK-8分析来研究MTHFD2、TYMS和MTR的改变如何影响肿瘤细胞生长。结果,MTHFD2或TYMS的过表达显著提高了肿瘤细胞的增殖率(图3g),而MTHFD2或TYMS过表达促进的肿瘤细胞生长因MTR的过表达而降低(图3g)。他们进一步评估了PRKDC过表达(OE)条件下MTHFD2、TYMS和MTR对肿瘤细胞增殖的影响。为此,他们分别将MTHFD2、TYMS和MTR过表达或敲低载体转染到PRKDC过表达的HMCB细胞系(PRKDC-OE-HMCB)中,并检测肿瘤细胞的增殖率。结果显示,与含有 MTHFD2-OE 或 TYMS-OE 的 HMCB 相比,过表达 MTHFD2 或 TYMS 的 PRKDC-OE 细胞表现出显著增加的肿瘤细胞增殖(图3h)。
蛋氨酸循环利用一碳单位进行甲基化,它与叶酸循环竞争,而叶酸循环利用源自丝氨酸的一碳单位进行 DNA 合成。因此,他们假设 MTR 的缺失可能会解开叶酸循环和蛋氨酸循环之间的联系,导致 DNA 合成池中一碳单位富集。与此假设一致的是,细胞中 MTR 的敲低导致叶酸循环代谢物(如丝氨酸、甘氨酸和 dTMP)的产生增加,并且 dTMP 与 dUMP 的比率增加(图3i)。相反,MTR 敲低细胞中的蛋氨酸循环受损,蛋氨酸和 SAM 的产生以及 SAM/SAH 比率降低(图3i)。黑色素瘤组织中丝氨酸、甘氨酸、dTMP 和 dTMP/dUMP 比率的增加以及蛋氨酸和 SAM 水平以及 SAM/SAH 比率的降低也验证了这一假设(图3j)。敲低 MTR 后,DNA 合成速率显著增加,并且在 MTHFD2 或 TYMS 过表达细胞中 G1 期细胞的数量显著减少(图3k)。通过使用异种移植瘤模型,他们发现MTR缺失和MTHFD2过表达诱导的异种移植瘤生长可以通过施加丝氨酸代谢抑制剂NCT503(该抑制剂靶向关键丝氨酸代谢酶PHGDH)来消除(图3l-m)。这些数据表明,MTR缺失结合MTHFD2上调导致一碳单位富集,并促进PRKDC-MXD3/ S57诱导的黑色素瘤肿瘤生长(图3n)。
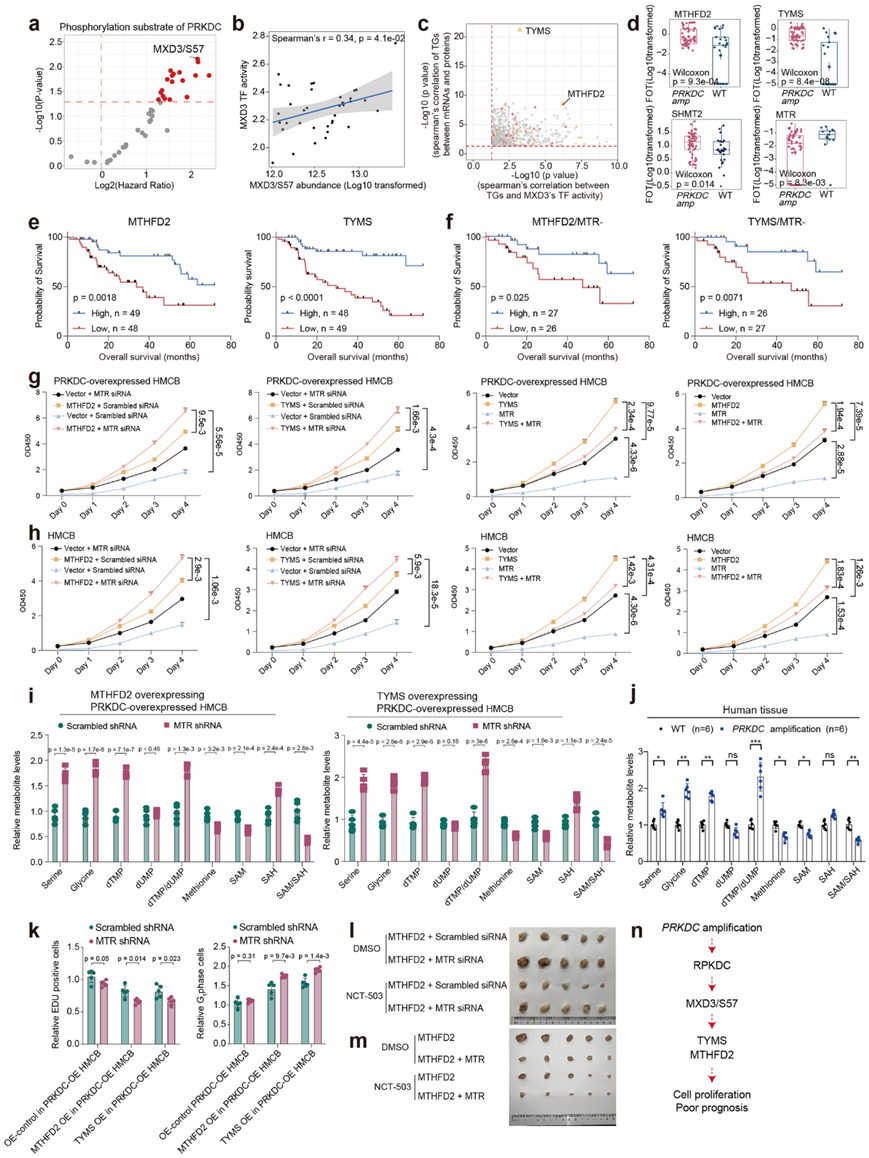
图3. 异常的叶酸代谢平衡导致黑色素瘤的肿瘤发展。
(a) 火山图显示了 PRKDC 磷酸化底物的丰度,可预测黑色素瘤中的 OS。(b) 黑色素瘤中 MXD3 的 TF 活性和 MXD3/S57 丰度的 Spearman 相关性。(c) MXD3 的 TF 活性和 MXD3 的 TG 蛋白表达的 Spearman 相关性(x轴);MXD3 的 TG 的 mRNA 表达和蛋白表达的 Spearman 相关性(y轴)。(d) MTHFD2、TYMS、SHMT2 和 MTR 的表达情况。(e-f)生存分析。 (g-h) HMCB 细胞的增殖情况。(i) HMCB 细胞的代谢情况。(j) 本研究队列中含有PRKDC扩增子和 WT 样本的患者的黑色素瘤组织中的代谢情况。(k) HMCB细胞的DNA合成情况和G1期细胞的数量分析。(l-m) 异种移植肿瘤模型分析。(n) RKDC-MXD3 信号通路的激活与一碳单位富集相结合导致黑色素瘤肿瘤生长的图示。
04
黑色素瘤的蛋白质组学亚型
鉴于肿瘤间的异质性,进行分子分型很重要。由于蛋白质组学数据直接反映了细胞的功能,他们根据137个黑色素瘤的蛋白质表达水平进行了共识聚类;随后,他们确定了三个亚组(S-I、S-II和S-III)(图4a)。值得注意的是,生存分析显示蛋白质组亚组在OS方面存在显著差异(图4b)。亚组特异性通路富集分析揭示了三个蛋白质组亚型之间的不同特征。在三个亚组中,SI 的特点是调节 ECM-受体相互作用和粘着斑(称为 ECM 亚型)。LAMA3、LAMB3 和 COL3A1 等蛋白质在 SI 亚型中占主导地位。此外,S-II 以 VEGF 信号通路、上皮细胞信号通路和 Hippo 信号通路为特征(称为血管生成亚型)。与此一致,包括PAK1、RAC1和PLCG1在内的蛋白在该亚型中过表达。此外,S-III 富集在核苷酸切除修复和碳代谢中(称为细胞增殖亚型)。在该亚型中,细胞增殖的关键调节因子(例如CDK4)的表达增加(图4a)。
值得注意的是,S-II 和 S-III 亚型均与不良预后相关,于是他们评估了PRKDC的表达(图2e)。与 SI 亚型相比,S-II 和 S-III 均显示 PRKDC 表达增加(图4c)。PRKDC扩增,S-III的CDK4扩增频率也增加(S-I为18%,S-II为12%,S-III为37%)(图4d-e)。此外,S-II 具有更高的ROCK2扩增频率(SI 中 14%、S-II 中 59% 和 S-III 中 31%),且CDK4扩增和ROCK2扩增互相排斥(图4d-e)。S-III 也表现出明显更高的多基因增殖评分(MGPS),表明在蛋白质组学水平上细胞增殖的富集增强(图4f)。CDK4 的激酶活性在 S-III 亚型中也明显更高,并与 MGPS 呈正相关,表明 CDK4 可能通过磷酸化促进 S-II 亚型中的肿瘤细胞增殖(图4g-h)。他们检测了 CDK4 抑制剂对细胞活力的影响,S-III 疾病患者的 PDC 对 CDK4 抑制剂 (Palbociclib) 更敏感,IC50值明显较低(中位 IC 50:PDC_S-II 中为 7.88 μM,PDC_S-III 中为 57.07 μM)(图4i)。
有趣的是,与仅CDK4扩增的患者相比,CDK4和PRKDC扩增患者的CDK4 激酶活性和 MGPS 明显更高(图4j)。与此同时,MGPS 在同时存在CDK4和PRKDC扩增的患者中也显著升高(图4k)。他们假设 PRKDC 可以磷酸化 CDK4 并增强其激酶活性,研究了 PRKDC 的磷酸化底物,发现 CDK4 在 T172 处的磷酸化与 PRKDC 激酶活性、CDK4 激酶活性和 MGPS 呈正相关(图4l)。IHC 染色进一步证实了 S-III 中 CDK4/T172 磷酸化的升高(图4m)。
为了证实PRKDC-CDK4级联在促进肿瘤细胞增殖方面的作用,他们收集了患者来源的PDC进行进一步分析(PDC_ PRKDC Amp & CDK4 Amp:属于S-III且同时具有PRKDC扩增和CDK4扩增的患者;PDC_WT:属于没有PRKDC扩增和CDK4扩增的患者)。他们对PDC_ PRKDCAmp & CDK4Amp和PRDC_WT的蛋白质组进行了比较分析。结果发现,在PDC_ PRKDCAmp & CDK4 Amp中,在增殖通路富集的蛋白质水平显著升高(图4n-o)。PRKDC 和 CDK4 抑制剂的联合使用最显著地降低了 PDC_ PRKDC Amp和CDK4 Amp细胞的增殖,表明 PRKDC 可以增强 CDK4 促进 S-III 亚型肿瘤细胞增殖的能力(图4p)。总之,这些数据表明 S-III 的细胞增殖特征是由CDK4扩增驱动的,并且可以通过 PRKDC 抑制剂进一步增强。CDK4 和 PRKDC 抑制剂的联合使用可以使 S-III 患者在临床上受益(图4q)。
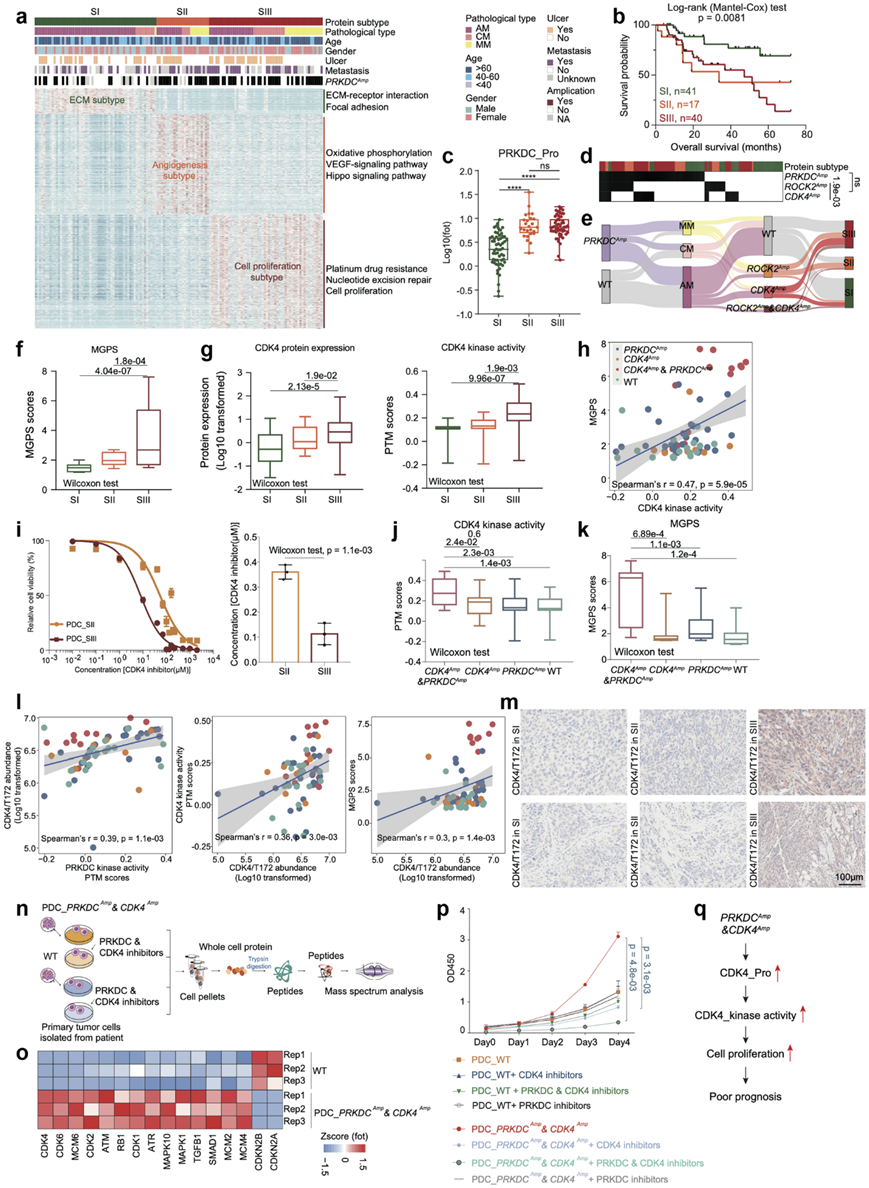
图4. 原发性黑色素瘤的蛋白质组亚型。
(a) 热图显示了 137 名黑色素瘤患者的临床信息和PRKDC扩增子的频率。(b) 三种蛋白质组亚型与黑色素瘤患者临床结果的关联。(c) 箱线图显示了三种蛋白质组亚型中 PRKDC 的蛋白质表达 。(d) 热图显示了三种蛋白质组亚型中PRKDC、CDK4、ROCK2的扩增频率。(e) 桑基图显示黑色素瘤三种病理亚型和三种蛋白质组亚型中PRKDC、CDK4和ROCK2的扩增频率。(f) 箱线图显示三种蛋白质组亚型中的 MGPS 评分。(g) 箱线图显示三种蛋白质组亚型中 CDK4 的蛋白表达和激酶活性。(h) 黑色素瘤中 CDK4 激酶活性与 MGPS 评分的 Spearman 相关性。 (i) 剂量反应曲线。(j) 箱线图显示了本研究队列中含有CDK4扩增子和PRKDC扩增子、仅PRKDC扩增子或仅CDK4扩增子和 WT 样本的患者的 CDK4 PTM 评分。(k) 箱线图显示了本研究队列中含有CDK4扩增子和PRKDC扩增子、仅PRKDC扩增子或仅CDK4扩增子和 WT 样本的患者的 MGPS 评分。(l) 黑色素瘤中PRKDC激酶活性与CDK4/T172丰度的Spearman相关性(左);黑色素瘤中CDK4/T172丰度与CDK4激酶活性的Spearman相关性(中);黑色素瘤中CDK4/T172丰度与MGPS评分的Spearman相关性(右)。 (m) 免疫组织化学分析。(n)实验流程。(o) 热图显示参与细胞周期的CDK4、CDK6等蛋白质表达在含有CDK4扩增子和PRKDC扩增子的黑色素瘤患者的PDC中上调。(p) PDC 增殖情况。(q) 说明PRKDC-CDK4 信号通路的激活与细胞增殖相结合导致黑色素瘤预后不良。
05
ROCK2 扩增促进原发性黑色素瘤的转移
值得注意的是,虽然 S-II 亚组和 S-III 亚组均与不良预后相关,但 S-II 亚组包含更高比例的转移性患者(图5a)。为了进一步分析与 S-II 亚组的转移性特征相关的可能细胞过程,他们首先比较了三个蛋白质组亚组之间的基因组改变,并确定ROCK2是唯一在 S-II 亚组中显示出明显更高扩增频率、mRNA 表达和蛋白质表达的 CAG(图5b)。从功能上讲,ROCK2 是一种参与血管生成和上皮细胞信号通路的激酶,并与肿瘤细胞转移有关。与此一致的是,TCGA 队列中发生转移的原发性黑色素瘤中 ROCK2 的 mRNA 表达和 GSVA 评分增加(图5c)。使用抗 ROCK2 抗体通过 IHC 染色进一步证实了转移性原发性黑色素瘤中 ROCK2 表达的升高(图5d)。根据这些研究,他们发现在具有ROCK2扩增子的患者中,调节细胞骨架、上皮细胞信号通路和黏附连接等通路活性显著升高(图5e)。生存分析显示,HMGB1 在 S118 处的磷酸化是与患者预后呈负相关的 ROCK2 相关磷酸位点中的关键磷酸位点之一(图5f)。称为血管生成核心调节因子的 PDGFRA 是唯一在 mRNA 和蛋白质水平上与 OS 呈负相关的 TG,这表明 HMGB1 可能通过促进肿瘤血管生成来驱动患者预后不良(图5g)。
为了验证从 ROCK2 到 HMGB1 的级联过程,他们使用 pCDH-ROCK2-copGFP 载体构建了稳定的 ROCK2 过表达 A375 细胞系 (ROCK2-OE-A375),并使用 pLKO.1-CMV-shROCK2-copGFP 敲低ROCK2 (ROCK2-KD-A375)。与对照细胞相比,ROCK2-OE-A375 细胞系表现出细胞迁移增加,而 ROCK2-KD-A375 细胞系表现出细胞迁移减少(图5h-i)。
他们研究了HMGB1对下游血管生成过程的影响,使用pCDH-HMGB1-copGFP载体构建了稳定的HMGB1过表达A375细胞系(HMGB1-OE-A375),并使用pLKO.1-CMV-HMGB1-copGFP敲低HMGB1(HMGB1-KD-A375),然后进行了RT-PCR以评估细胞(HMGB1-OE-A375,HMGB1-KD-A375,OE-Control-A375和KD-Control-A375)中血管生成相关蛋白的表达。结果显示,与血管生成有关的基因在HMGB1-OE-A375中的表达高于在OE-Control-A375中的表达。但和KD-Control-A375细胞相比,HMGB1-KD-A375细胞中血管生成过程相关基因表达降低(图5j),证实了HMGB1在促进血管生成中的调控作用。为进一步验证HMGB1,特别是磷酸化HMGB1对肿瘤细胞迁移的作用,他们构建了HMGB1-OE载体和HMGB1-S100-mutation-OE载体,转染到HMGB1-KD-A375稳定细胞株中,通过Transwell实验评估细胞迁移能力。和KD-Control-A375相比,HMGB1-KD-A375显著降低肿瘤细胞迁移能力。与KD-Control-A375相比,HMGB1-KD-A375显著降低肿瘤细胞迁移,而转染HMGB1-S100-mutation-OE载体后,细胞迁移能力的下降趋势保持不变(图5k-l),说明HMGB1 S100位点磷酸化对促进肿瘤细胞迁移起着至关重要的作用。
为了进一步证实ROCK2(激酶)-HMGB1 TF-PDGFRA TG级联促进肿瘤转移,他们构建了一个包括20例黑色素瘤患者的独立验证队列,并收集了匹配的原发性和转移性黑色素瘤肿瘤样本进行蛋白质组学和磷酸化蛋白质组学分析(图5m)。他们比较了匹配的原发性和转移性黑色素瘤肿瘤样本中ROCK2的表达和HMGB1的磷酸化。结果发现,在转移性样本中ROCK2的蛋白表达和HMGB1的磷酸化均升高(图5n)。
此外,为了验证 ROCK2 通过 HMGB1 磷酸化促进肿瘤转移的潜在因果关系,他们还收集了患者的 PDC(PDC_ ROCK2 Amp:属于ROCK2扩增的患者,PDC_WT:属于ROCK2未扩增的患者),并进一步进行了蛋白质组学和磷酸化蛋白质组学分析(图5o)。结果显示,参与血管生成的蛋白质水平,包括 ROCK2、PDGFRA、VEGFA 和 HMGB1,在 PDC_ ROCK2Amp中明显较高。此外,在磷酸化蛋白质组水平上,S100 处 HMGB1 的磷酸化是 ROCK2 最显著升高的磷酸化底物(图5p-q)。总的来说,这些数据表明,S-II 亚组中ROCK2扩增增高可能是血管生成增高的原因,并可能作为黑色素瘤转移的预测标志物(图5r)。具体而言,PRKDC扩增与CDK4扩增相结合增加了 S-II 中的细胞增殖,而ROCK2扩增增加了血管生成并促进了 S-III 中的黑色素瘤转移。
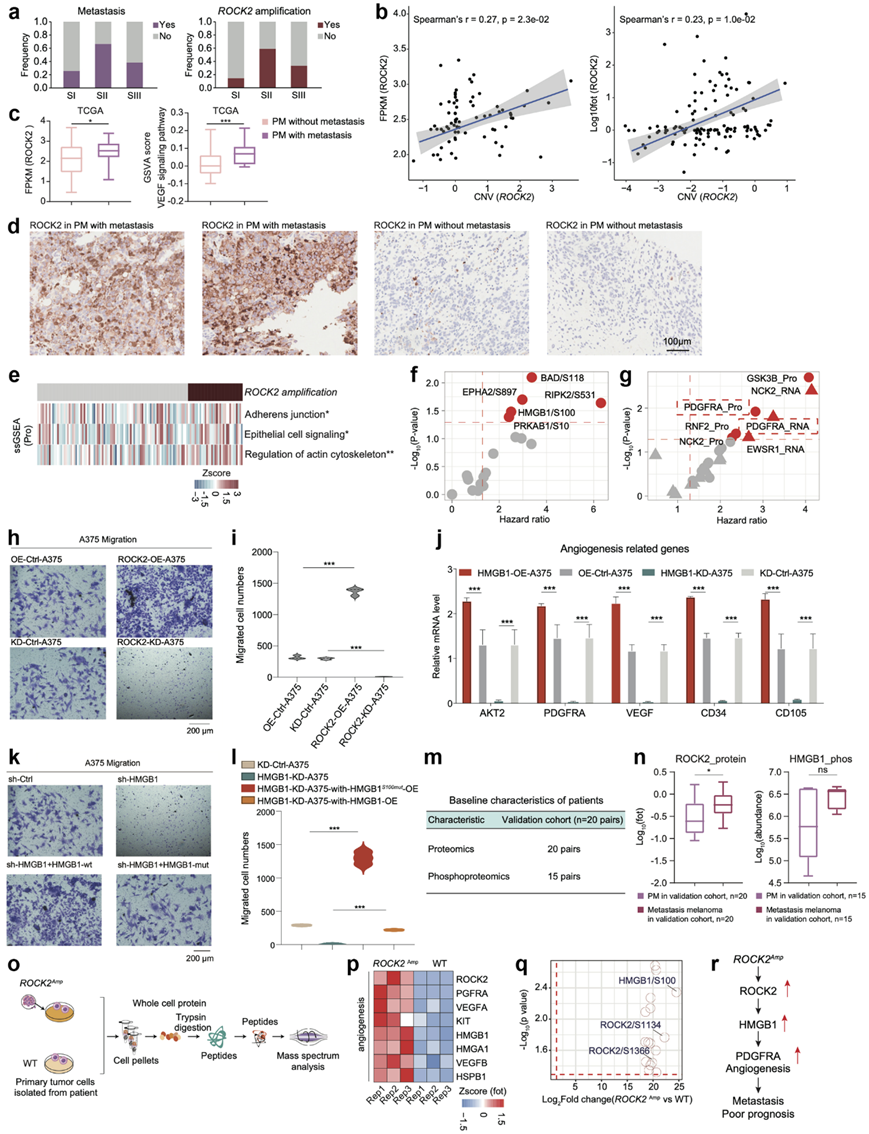
图5. ROCK2扩增促进黑色素瘤转移。
(a) 直方图显示转移和ROCK2扩增的频率。(b) 黑色素瘤中ROCK2拷贝数和ROCK2 mRNA 表达的Spearman 相关性(左);黑色素瘤中ROCK2拷贝数和ROCK2蛋白表达 的 Spearman 相关性 (右)。(c) 黑色素瘤中 ROCK2 的 mRNA 表达和 VEGF 信号通路的 GSVA 评分。(d) 免疫组织化学分析。(e) 热图描绘了在含有ROCK2扩增子的样本中显著升高的通路。(f) 火山图显示了预测黑色素瘤 OS 的磷酸位点的丰度。(g) 火山图显示了预测黑色素瘤 OS 的 HMGB1 的 TG 表达。(h) 通过transwell 证实了 ROCK2 对 A375 细胞迁移的影响。(i) 小提琴图显示不同处理下迁移的 A375 细胞计数。 (j) 箱线图显示了 OE-Control-A375、OE-HMGB1-A375、KD-Control-A375 和 HMGB1-KD-A375 中血管生成相关基因的表达水平。(k) 通过 transwell 确认了 HMGB1 对 A375 细胞迁移的影响。(l) 小提琴图表示不同处理下迁移的 A375 细胞数。(m) 验证队列 1 中患者的基线特征。(n) ROCK2 的蛋白质表达和 HMGB1 的磷酸化丰度。(o)实验流程。(p) 热图显示 ROCK2、VEGFRA、HMGB1 等参与血管生成的蛋白质表达在含有ROCK2扩增子的黑色素瘤患者的 PDC 中上调。(q) 火山图显示具有ROCK2扩增子的黑色素瘤患者中磷酸化显著上调。 (r) 示意图总结了 ROCK2 对血管生成和通过 HMGB1 促进黑色素瘤转移的级联调控作用。
06
具有不同生物学和临床特征的免疫亚群
他们基于 RNA-seq 数据进行了 xCell 分析,以推断肿瘤微环境中不同细胞类型的相对丰度。基于推断的细胞比例的共识聚类有助于识别具有不同免疫特征的三组肿瘤(S1-S3)(图6a)。生存分析表明,免疫亚组在 OS 方面存在显著差异,这表明不同类型的免疫细胞浸润可导致不同的预后结果(图6b)。此外,他们观察到<50%的AM分布在S1亚型中,而大多数MM分布在S3亚型中,且S3亚型中女性患者的比例明显高于其他两个亚型(图6c)。3个亚型中,S1以CD4 + T细胞和CD8 + T细胞为特征(称为T细胞亚型),CD8A、PDCD1、CD247、CD274、CD3D等mRNA在S1亚型中优势表达。ECM受体相互作用、MAPK信号通路等通路在S1亚型中优势表达(图6d)。S2以巨噬细胞特征为特征(称为TAM亚型),CD68、CD163、CD63等mRNA在该亚型中过表达。参与 VEGF 信号通路、ErbB 信号通路和溶酶体的蛋白质在该亚型中过多表达(图6d)。此外,S3 富含 γδT 细胞和 NKT 细胞特征,并且 IL-17 的 mRNA 表达在该亚型中增加(称为 IL-17 分泌亚型)。细胞周期、嘧啶代谢和叶酸单碳循环等通路在 S3 亚型中主要表达(图6d)。使用 CD8、CD163 和 IL17 进行 IHC 染色进一步证实了 S1 亚型中 CD8 的富集、S2 亚型中巨噬细胞的富集以及 S3 亚型中 γδ T 细胞的富集(图6h)。
重要的是,与 S2 和 S3 相比,S1 亚型包含更多的 AM 患者,这意味着相当数量的 AM 患者可能表现出免疫细胞浸润升高(图6e)。此外,由于观察到 S1 亚型中PD-L1 表达升高和 CD8 + T 细胞高富集(图6f-g),他们假设 S1 亚型患者对免疫治疗的敏感性可能高于其他亚型患者。为了阐明这种现象背后的可能机制,他们比较了三种免疫亚型的分子特征,发现 MAPK 信号通路和 NFκB 信号通路与 CD8 + T 细胞特征评分呈正相关。通过比较三个免疫亚组之间的激酶活性,他们发现 MAPK7(ERK5)的激酶活性是与 CD8+ T 细胞特征评分相关的最高激酶(图6i)。他们评估了 NFκB2 的 TF 活性,发现 NFκB1/2 的 TF 活性也与 CD8+ T 细胞的富集评分呈显著正相关性(图6j)。参与 T 细胞募集的细胞因子/趋化因子(如 CXCL4 和 CXCL9)在 S1 亚组中也升高(图6k)。
总之,这些结果表明 MAPK7 激酶活性升高可能增强 NFκB2 的 TF 活性,进而增加细胞因子表达和 CD8+ T 细胞的募集(recruitment)。因此,S1 亚型患者可能从免疫疗法中受益。
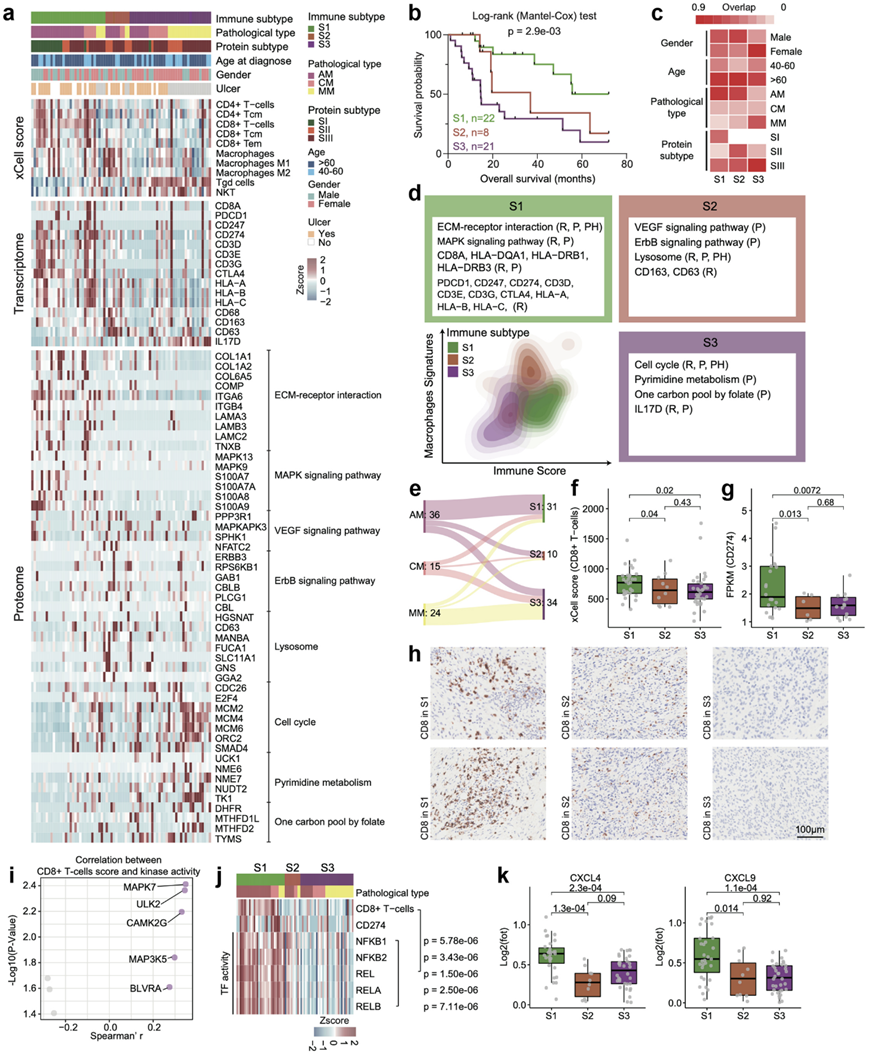
图6. 黑色素瘤的免疫状况。
(a) 热图显示了 75 名黑色素瘤患者的三个免疫组中选定的单个 mRNA/蛋白质和通路的细胞类型组成和活性。(b) 三种免疫组与黑色素瘤患者临床结果的关联。(c) 热图显示了免疫聚类(列)与性别、年龄、蛋白质组亚型和不同组织学类型的比较。 (d) 基于不同免疫组的巨噬细胞(y轴)和免疫评分(x轴)的二维密度轮廓图。(e) 桑基图显示了不同组织学类型的免疫聚类之间的比较。 (f) 三个免疫聚类中CD8 + T 细胞的 xCell 评分。(g) 箱线图显示三个免疫聚类中 CD274 的 mRNA 表达。(h) 三个免疫组中 CD8 的 IHC 。(i) 火山图显示了激酶活性与 CD8 + T 细胞的 xCell 评分显著相关的激酶。(j) 热图显示 NFκB 家族的 TF 活性与 CD8 + T 细胞的 xCell 评分呈正相关。(k) 箱线图显示了三个免疫组中 CXCL4 和 CXCL9 的蛋白质表达。
07
鉴定与黑色素瘤免疫治疗反应相关的蛋白质标记物
为了进一步说明 MAPK7 与黑色素瘤免疫治疗效果之间的潜在关联,他们构建了一个独立的验证队列,其中包括 27 名接受抗 PD1 治疗的 IV 期黑色素瘤患者(18 名 AM、6 名 CM、2 名 MM)。根据患者对治疗的反应将患者分组,其中 15 名应答者(包括部分和完全反应;n = 15)和 12 名无应答者(包括病情稳定和病情进展;n = 12)(图7a)。收集治疗前的肿瘤样本,并进行蛋白质组学和磷酸化蛋白质组学分析。
他们首先应用反卷积分析,并比较了应答者和无应答者之间的免疫细胞富集情况。结果显示,CD4+ T 细胞和 CD8+ T 细胞特征在应答者中富集(图7b)。利用抗 CD8 抗体的 IHC 染色也证实了应答者中 CD8 的富集(图7c)。与此一致的是, Harel 62和 Beck 63在之前的两项研究中也证实了应答者中 CD8+ T 细胞的富集(图7d)。此外,他们对应答者和无应答者进行了比较蛋白质组学分析,结果与 S1 免疫亚群的分子特征一致,应答者中升高的蛋白质富集的通路主要是 MAPK 信号通路和免疫相关通路,而无应答者则以代谢蛋白相关通路为主(图7e)。他们还比较了有反应者和无反应者之间的激酶活性。结果显示,MAP 激酶 MAPK7 (ERK5)、MAPK15 (ERK7)、MAPK11 和 MAP3K14 在应答者中高表达,而 DNA 修复相关激酶 PRKDC (DNAPK)、CSNK1A1 (CK1A)、CSNK2A2 (CK2A2) 和 DYRK1A 在无应答者中高表达(图7f)。
重要的是,为了验证 MAPK7-NFκB2 级联的激活是否导致参与募集 T 细胞的细胞因子表达增加,他们进行了相关性分析,并观察到细胞因子/趋化因子如 CCL5、CXCL4 和 CCL4 与 MAPK7 的激酶活性和 NFκB2 的蛋白表达呈正相关(图7g-h)。同时,他们构建了 MAPK7 过表达、MAPK7 敲低的 A375 细胞,用 MAPK7 抑制剂(XMD8-92)处理 A375 细胞,并使用野生型 A375 细胞作为对照。他们将这些细胞与 CD8 T 细胞共培养,并收集上清液进行蛋白质组学分析(图7i)。结果发现,在过表达MAPK7的A375细胞培养上清中,参与募集T细胞的细胞因子如CCL4、CCL8、CXCL4等显著上调(图7j),而在敲低MAPK7的A375细胞培养上清中,参与募集IL-17 T细胞的细胞因子/趋化因子(包括CCL2/6/7/13/14等)发生显著上调(图7j),同时,ELISA检测发现CD8 + T细胞与过表达MAPK7的A375细胞共培养后发生显著活化(图7k-l),这些结果证明了MAPK7在募集和激活CD8 + T细胞方面的作用,并有助于免疫治疗。
为了确定可能促进抗PD-1免疫疗法疗效的潜在可用药靶点,他们重点关注了在无应答组表达升高的激酶。PRKDC是无应答者中表达升高最显著的激酶。公开队列进一步证实了这一现象(图7m)。因此,PRKDC的蛋白质表达与MAPK7的蛋白质表达呈负相关(图7n)。为了评估靶向PRKDC增强免疫疗法疗效的潜力,他们在各种处理条件下(MAPK7敲低的A375细胞、用PRKDC抑制剂处理的A375细胞、用PRKDC抑制剂处理的MAPK7敲低的A375细胞和用PRKDC抑制剂处理的MAPK7过表达的A375细胞)将T细胞与A375细胞共培养,随后进行了ELISA 检测,发现 MAPK7 过表达的 A375 细胞和 PRKDC 抑制剂显著地激活了CD8+ T 细胞(图7o)。这些结果强调了 PRKDC 表达降低可以显著激活 CD8+ T 细胞。
基于这些结果,他们进一步利用MAPK7-KD-A375稳定细胞系构建异种移植黑色素瘤小鼠模型,并以KD-Control-A375细胞系作为对照。连续检测异种移植肿瘤三周,然后用PD-1抑制剂或PRKDC(NU7441)和PD-1抑制剂联合治疗或不进行治疗。在三周的治疗期间,连续检测肿瘤大小。与移植了KD-Control-A375细胞的小鼠相比,移植了MAPK7-KD-A375细胞的小鼠对PD-1治疗不敏感(图7p),表明MAPK7表达降低显著降低了小鼠对PD-1治疗的反应。与此结果一致的是,他们观察到与移植了KD-Control-A375细胞的小鼠相比,移植了MAPK7-KD-A375细胞的小鼠中与T细胞募集相关的蛋白质(例如CCL5和CCL3)的表达明显较低(图7q)。此外,对于移植了MAPK7-KD-A375细胞的小鼠,与单独使用PD-1治疗相比,PRKDC与PD-1抑制剂的组合显著降低了肿瘤生长(图7r),证实了使用PRKDC抑制剂与抗PD-1抗体联合用于黑色素瘤治疗的潜力。
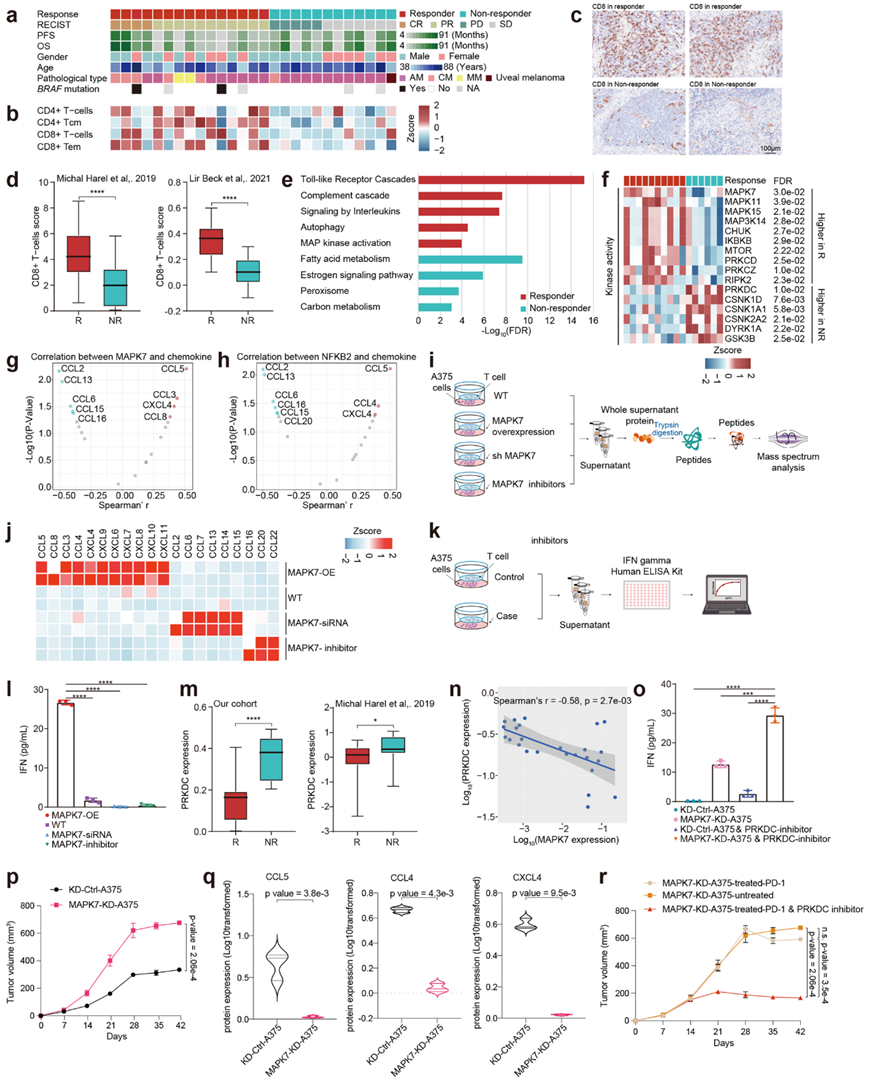
图7. 鉴定黑色素瘤对免疫疗法有反应的蛋白质标记物。
(a) 热图显示了抗 PD-1 队列的临床信息。(b) 热图显示了基于使用 xCell 分析的免疫特征。(c) 免疫组织化学分析。(d) CD8+ T 细胞的 xCell 评分。(e)上调的通路分析。(f) 热图显示了应答者和无应答者中激酶活性上调的激酶。(g) 火山图显示蛋白表达与 MAPK7 激酶活性显著相关的趋化因子。(h) 火山图显示蛋白表达与 NFKB2 蛋白表达显著相关的趋化因子。(i) 实验流程。(j) 热图说明不同处理条件下 A375 细胞中趋化因子的蛋白质表达。(k) 实验流程。(l) 直方图显示不同处理条件下 A375 细胞与 T 细胞培养上清液中的 IFN 浓度。(m) PRKDC 的蛋白质表达分析。(n) 黑色素瘤中 MAPK7 蛋白表达和 PRKDC 蛋白表达的 Spearman 相关性。 (o) 直方图显示不同处理条件下 A375 细胞与 T 细胞培养上清液中的 IFN 浓度。(p) 肿瘤生长曲线。(q) 箱线图显示 KD-MAPK7-A375 肿瘤和 KD-Ctrl-A375 肿瘤之间 CCL5、CCL4 和 CXCL4 的蛋白质表达。(r) 肿瘤生长曲线。
+ + + + + + + + + + +
结 论
本项研究的多组学分析显示,PRKDC扩增是黑色素瘤的预后分子,PRKDC扩增的顺式效应可能通过激活 DNA 修复和叶酸代谢通路导致肿瘤增殖。基于蛋白质组的原发性黑色素瘤分层定义了三种与预后相关的亚型,即 ECM 亚型、血管生成亚型(转移率高)和细胞增殖亚型,这为利用针对特定黑色素瘤亚型的特定靶向疗法提供了新的理论依据。结合独立的抗 PD-1 治疗队列的进一步分析表明,MAPK7-NFKB 信号通路的上调可能促进 T 细胞募集并增加患者对免疫治疗的敏感性。相反,PRKDC 可能通过促进黑色素瘤细胞中的 DNA 修复来降低黑色素瘤患者对免疫治疗的敏感性。
+ + + + +



