

 English
English文献解读|Nat Commun(14.7):多生物群落分析识别出溃疡性结肠炎和克罗恩病中不同的肠道微生物特征及相互作用
✦ +
+
论文ID
原名:Multi-biome analysis identifies distinct gut microbial signatures and their crosstalk in ulcerative colitis and Crohn’s disease
译名:多生物群落分析识别出溃疡性结肠炎和克罗恩病中不同的肠道微生物特征及相互作用
期刊:Nature Communications
影响因子:14.7
发表时间:2024.11.27
DOI号:10.1038/s41467-024-54797-8
背 景
炎症性肠病 (IBD) 中的细菌组已得到广泛研究,其特点是细菌组多样性和短链脂肪酸 (SCFA) 产生菌减少,以及几种致病菌的富集,包括肠杆菌科植物和屎肠球菌。就细菌基因功能而言,最初从 CD 患者中分离出的粘附侵袭性大肠杆菌(AIEC) 能够穿透粘液层并通过 1 型菌毛粘附附着于肠上皮细胞系,表明这种细菌功能在诱导肠道炎症中起着至关重要的作用。此外,与回肠CD 相关的大肠杆菌往往具有多种耐药性。一组具有独特抗生素耐药基因 (ARG) 的肺炎克雷伯菌菌株与 IBD 的病情恶化和严重程度有关。这些发现意味着肠杆菌科的几种菌种与 IBD 的发病机制有关。然而,溃疡性结肠炎 (UC) 和克罗恩病 (CD) 中肠道微生物组的综合多界相互作用仍未得到充分研究。
实验设计
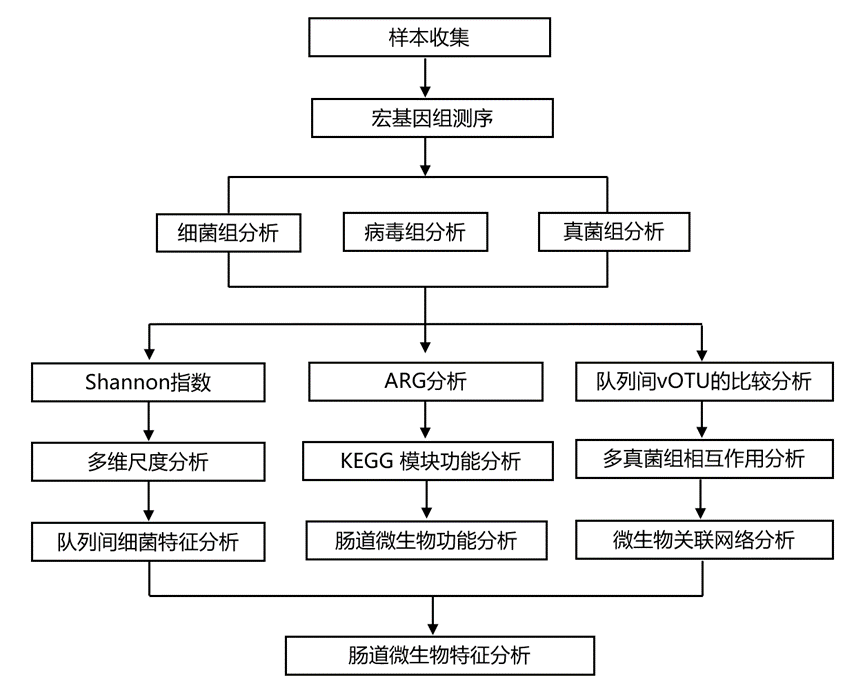
结 果
01

UC 和 CD 中的共生菌和致病菌的细菌组特征不同
研究团队对日本 4D(疾病、药物、饮食、日常生活)队列(年龄 16-89 岁,59% 为男性)中的 111 名 UC 患者、31 名 CD 患者和 540 名健康对照者的粪便样本进行了宏基因组测序。与对照组相比,日本 4D 队列中 UC 和 CD 患者的细菌组 α 多样性显著降低,CD 患者的 α 多样性显著低于 UC 患者(图 1a)。多维尺度 (MDS) 分析显示,三组肠道细菌组的 β 多样性存在显著差异(图 1b)。
通过年龄、性别和 BMI 调整的回归模型,与对照组相比,IBD、UC 和 CD 患者中分别有 263、214 和 133 种细菌发生了显著变化,特别是IBD 中双歧杆菌属(短双歧杆菌、长双歧杆菌和齿双歧杆菌)、肠球菌属(屎肠球菌和粪肠球菌)和唾液链球菌的显著富集,以及产生 SCFA 的细菌如粪杆菌、真杆菌属(E. rectale 和 E. ventriosum)和Anaerostipes hadrus的减少。没有任何属于古菌的物种(例如Methanobrevibacter smithii)与 IBD 有显著关联。
为了验证日本 4D 队列中结果的可重复性,他们将回归模型中每种细菌的系数与外部队列的系数进行了比较。分析显示所有队列都存在显著的正相关性。日本 4D 和美国队列在 IBD、UC 和 CD 中的细菌特征相似(图 1c),其他队列中的细菌特征也相似。细菌分析表明,在日本 4D 队列中,屎肠球菌在 UC 和 CD 中显著富集,而大肠杆菌与 CD 特异性相关(图 1d)。这一发现表明 UC 和 CD 可能具有不同的致病菌,他们接下来进行了更深入的分析以识别 ESKAPE 病原体,这些病原体是 WHO 确定的已知可导致多重耐药院内感染的病原体。结果显示,肺炎克雷伯菌、金黄色葡萄球菌、鲍氏不动杆菌和铜绿假单胞菌在 CD 中显著增加,但在 UC 中没有增加(图 1e)。粪肠球菌在 UC 和 CD 中均显著富集。由于本研究队列中在 CD 中富集的 ESKAPE 致病菌是已知的多重耐药细菌,因此 CD 中的多重耐药细菌可能比 UC 患者中更多。
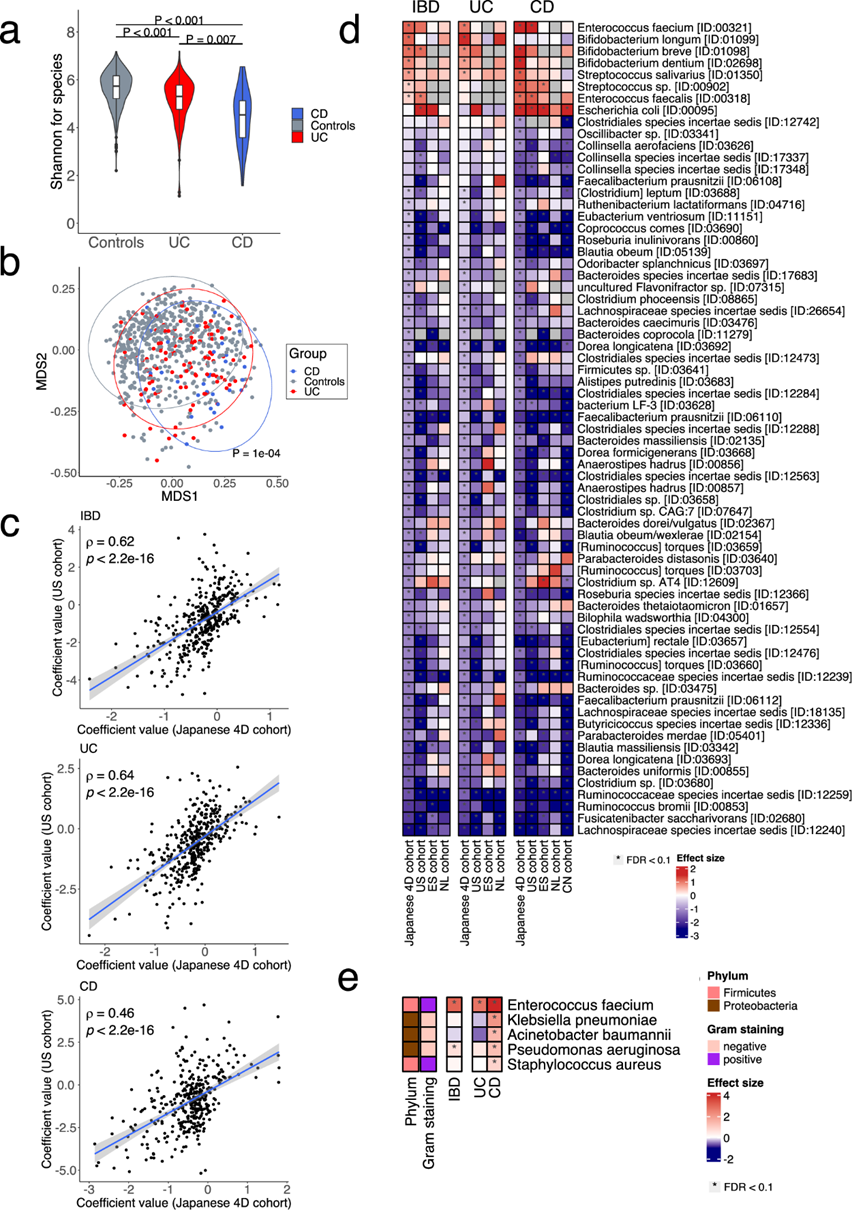
图1. UC 和 CD 的肠道细菌组特征。(a) 小提琴图显示对照组、溃疡性结肠炎 (UC)和克罗恩病 (CD)的肠道细菌组的香农指数。(b) 多维尺度 (MDS) 图显示样本的相似性。(c) 散点图显示在日本队列(JP, x轴)和美国队列(US,y轴)中确定的炎症性肠病 (IBD)、UC 和 CD 的细菌物种特征的系数值。(d) 热图显示了与日本 4D 队列、美国 (US)、西班牙 (ES)、荷兰 (NL) 和中国 (CN) 队列中的健康对照相比,IBD 患者中富集和减少的物种。(e) 热图显示了与日本 4D 队列中的健康对照相比,IBD、UC 和 CD 中 ESKAPE 病原体富集和减少的物种。
02
耐药性分析揭示了大肠杆菌与 CD 中的抗生素外排泵之间存在特定关联
为了研究微生物组的抗生素耐药性潜力,他们对日本 4D 队列的耐药组(resistome)中的 ARG 进行了分析,并在各组之间进行了比较。CD 中肠道耐药组的 α 多样性略高于对照组和 UC,但不显著(图 2a)。MDS 分析显示三组之间的 β 多样性存在显著差异,表明耐药组的组成不同(图 2b)。CD 中的 ARG 数量及其总丰度明显高于 UC 或对照组(图 2c-d),这表明 CD 患者的肠道共生菌更有可能拥有 ARG。与对照进行比较分析,发现在 IBD、UC 和 CD 中分别有 26、20 和 77 种 ARG 发生显著变化。具体来说,外排泵(efflux pump)(如acrB、emrR和acrA)在 CD 中显著富集,而在 UC 中则没有显著变化。外排泵在 CD 中的显著富集表明它们导致了多重抗生素耐药细菌的出现。
接下来,他们检测了ARGs与细菌种类之间的相关性,发现在UC和CD中富集的msrC与屎肠球菌呈正相关,而屎肠球菌也富集在UC和CD中(图 2e)。值得注意的是,在CD中特异性富集的38种抗生素外排泵与大肠杆菌和肺炎克雷伯菌呈正相关。有趣的是,这些物种也与样本中检测到的ARGs总数呈现出显著的正相关性(图 2e),表明CD肠道中存在多重耐药细菌。此外,与相关性分析一致,综合抗生素耐药性数据库 (CARD) 表明肠杆菌科细菌具有这些 ARG。综上所述,这些研究结果表明,与健康人相比, IBD 患者感染多重抗生素耐药病原体(特别是大肠杆菌和肺炎克雷伯菌)的风险更大。

图2. UC 和 CD 的抗性基因组特征及其与具有抗生素抗性基因的物种的相关性。
(a) 小提琴图显示对照组、溃疡性结肠炎 (UC)和克罗恩病 (CD)的肠道抗药性的香农指数。(b) 多维尺度 (MDS) 图显示样本的相似性。(c) 箱线图显示在对照组、UC和 CD中发现的抗生素抗性基因 (ARG) 的数量 。(d) 箱线图显示对照组、UC和 CD中 ARG 的总丰度值。(e)热图描绘了与健康对照 (HC) 相比,UC 和 CD 中细菌种类与 ARG 之间的相关性。
03
IBD 中存在着独特的微生物功能,可以诱发炎症并抵抗抗菌防御
基于 KEGG 模块 (MO) 水平的功能分析发现,与对照组相比,IBD、UC 和 CD 患者中分别有 336、322 和 341 个 MO 发生了显著变化。具体来说,C5 异戊二烯类生物合成(萜类生物合成的甲羟戊酸途径)和阳离子抗菌肽 (CAMP) 抗性在 UC 和 CD 中均显著 (FDR < 0.1) 富集,而几个双组分调节系统则显著耗尽(图 3a-b)。在 UC 中,负责寡糖基转移酶 N-糖基化的 MO 明显减少,而在 CD 中,肝素、软骨素和硫酸皮肤素等糖胺聚糖的降解明显减少(图 3a-b)。
接下来,他们重点研究了 KEGG 直系同源物(KO)水平的肠道微生物功能,并分别鉴定出与对照组相比在 IBD、UC 和 CD 患者中发生显著变化的 3637、3347 和 3995 个 KO。与基于 MO 的结果一致(图 3a-b),参与萜类骨架生物合成的 KO 在 UC 和 CD 中均显著富集(图 3c)。负责 N-糖生物合成(K07252 和 K07151)和糖胺聚糖降解的 KO 在两种疾病中均显著减少(图 3d-e)。这些发现表明细菌糖代谢在 UC 和 CD 中下调。同时,一些属于 CAMP 抗性的 KO 在两种疾病中均显著增加。值得注意的是,CAMP 抗性基因(例如,属于 OmpR 家族的基因)在 CD 中特异性富集,但在 UC 中没有(图 3f)。IBD 中存在抵抗抗菌防御的独特机制,并且参与该机制的物种在 UC 和 CD 之间明显不同。
他们研究了与病原体相关分子模式分子有关的微生物功能以及 AIEC 的潜在毒力因子,它们可以激活炎症信号级联,可能在 IBD 中发挥重要作用。鞭毛组装和脂多糖 (LPS) 生物合成的 KO 在 CD 中显著富集,并且这些 KO 与大肠杆菌和肺炎克雷伯菌呈正相关(图 3g)。此外,肽聚糖生物合成的KO在UC和CD中均显著富集,并且这些KO与唾液链球菌和副血链球菌呈正相关,而后者在UC和CD中也显著富集(图 3g)。至于AIEC毒力因子,他们检测了之前报道的参与AIEC致病机制的30个KO,发现其中12个KO在CD中显著富集。它们的功能主要包括粘附和侵袭肠上皮细胞(例如,1型菌毛)(图 3h)。与这些基因呈正相关的细菌种类包括大肠杆菌和肺炎克雷伯菌。这一发现表明大肠杆菌在 CD 中具有 AIEC 的毒性基因,与之前的研究一致。
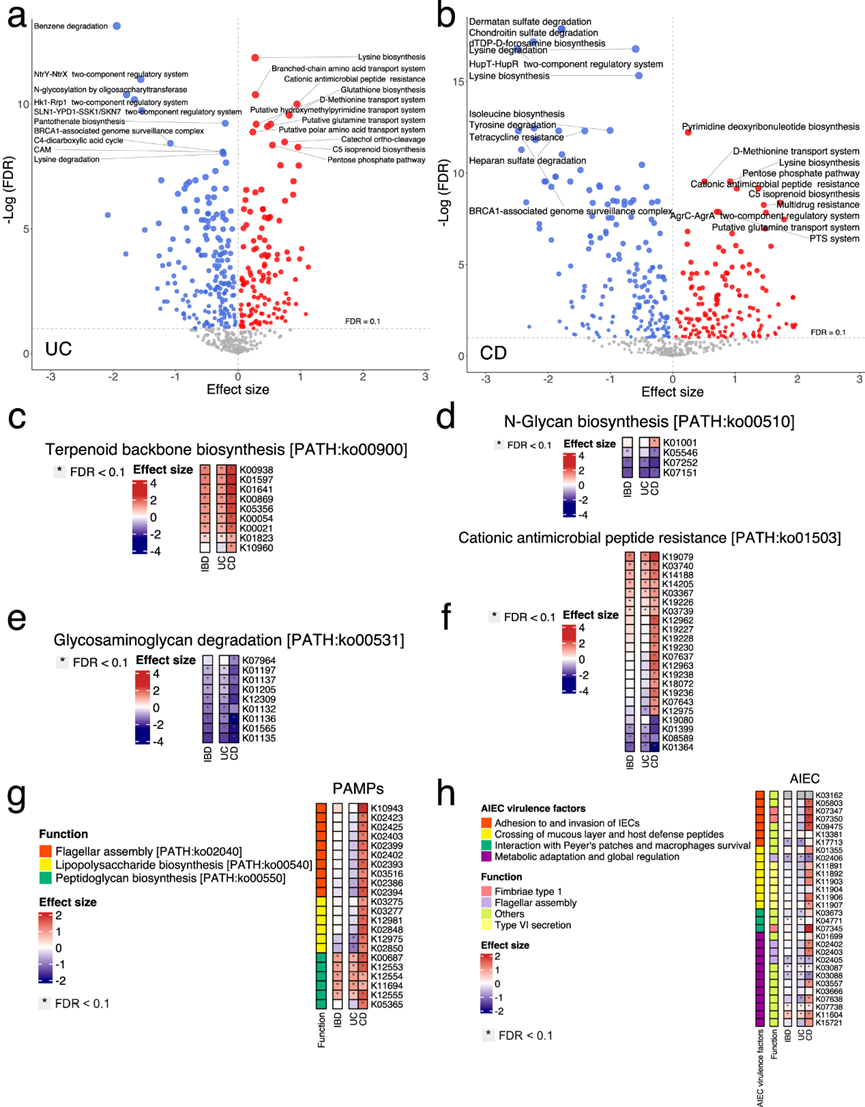
图3. UC 和 CD 的功能特征。
(a-b) 火山图显示了 KEGG 模块 (MO) 与溃疡性结肠炎 (UC)和克罗恩病 (CD)之间的关联。(c-f) 热图显示了功能结果。(g) 热图描绘了与健康对照 (HC) 相比,UC 和CD中 KO 关于病原体相关分子模式 (PAMP) 分子的系数值 (MaAsLin2)。(h) 热图描绘了 UC 和 CD 中粘附侵袭性大肠杆菌(AIEC) 毒力因子的 KO 系数值 (MaAsLin2) 与 HC 的比较。
04
IBD 患者的肠道病毒组编码致病蛋白发生了改变
接下来,他们通过将序列映射到病毒基因组目录来研究宏基因组中的病毒种类。与对照组相比,UC 和 CD 患者的病毒组的 Alpha 多样性明显较低,日本 4D 队列中 UC 的 Shannon 指数与 CD 相比明显下降(图 4a)。MDS 分析显示,三组病毒组的 β 多样性存在显著差异(图 4b)。三组之间溶原性噬菌体的总丰度没有显著差异,而预测仅感染一种物种的噬菌体(特异性噬菌体)的总丰度在 UC 和 CD 中均明显高于对照组(图 4c-d)。
通过对病毒操作分类单位 (vOTU) 的比较分析发现,在日本 4D 队列中,与对照组相比,IBD、UC 和 CD 患者中分别有 65、37 和 16 个 vOTU 发生了显著变化。关联性最强的 vOTU 的是UC 中的乳酸杆菌(vOTU1252) 和CD 中的链球菌(vOTU689)(图 4e)。同时,大肠杆菌的噬菌体 vOTU77在 CD 中特异性富集。在UC和CD中显著减少的vOTU中,最常见的预测宿主是Ruminococcus,其次是Blautia和Bacteroides,它们是UC和CD中减少的SCFA产生菌(图 4e)。细菌组和病毒组的一致改变意味着噬菌体组的丰富度和多样性降低可能是细菌组变化的次要原因。
比较日本 4D 和其他队列之间的病毒组特征(即每个 vOTU 的回归模型系数集),结果显示 IBD、UC 和 CD 呈显著正相关,表明结果的稳健性。特别是,预测宿主包括产 SCFA 细菌的噬菌体在所有队列中均持续减少,而 vOTU87、89、90 和 77 在美国、ES 和 CN 队列的 CD 患者中普遍富集(图 4e)。
值得注意的是,功能分析表明,在UC或CD中富集的7个噬菌体中有3个编码假定的致病基因。在CD中显著富集的vOTU89和vOTU90编码pagC基因(K07804),这是一种具有高度免疫原性的外膜蛋白。这些噬菌体的基因组大小分别约为45.9 kb和23.9 kb,与RefSeq中的参考噬菌体基因组没有序列相似性。它们是感染大肠杆菌的模板噬菌体,编码整合酶,并且一些基因与大肠杆菌基因组相似。此外,他们发现vOTU689编码rfb基因聚类,例如rfbB和rfbC(分别为K01710和K01790),这可能有助于增加O抗原多样性(图 4f)。该噬菌体的基因组大小约为 46.1 kb,CRISPR 间隔序列比对预测宿主为链球菌属。CD 中 vOTU89 和 vOTU90 的特异性富集在美国、ES 和 CN 队列中一致(图 4e)。这些结果表明,具有此类特异性基因的噬菌体的富集有助于 CD 患者细菌组的致病性。
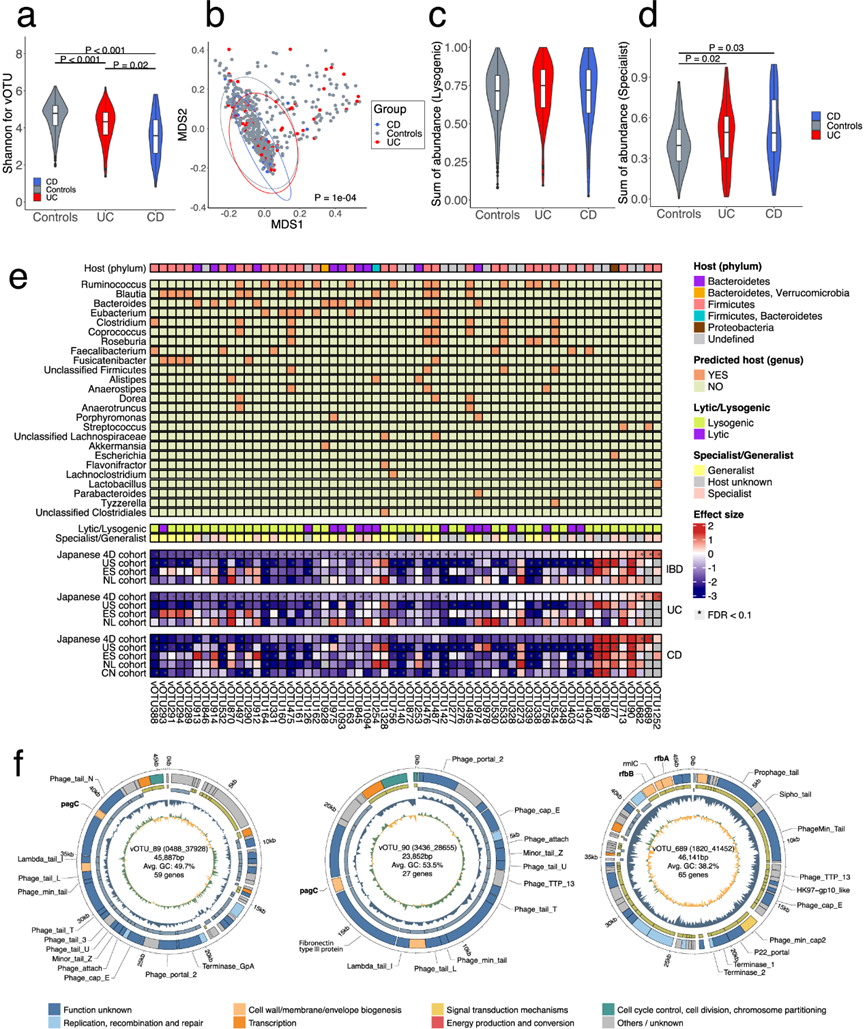
图4. UC 和 CD 的病毒组特征及其与预测宿主的相关性。
(a) 小提琴图显示对照组、溃疡性结肠炎 (UC)和克罗恩病 (CD)肠道病毒组的香农指数。(b) 多维尺度 (MDS) 图显示样本的相似性。(c) 箱线图显示对照组、UC和 CD中溶原性噬菌体的总丰度值 。(d) 箱线图显示了对照组、UC 组和 CD 组中专一噬菌体的总丰度值。(e) 热图显示了日本 4D 队列、美国 (US)、西班牙 (ES)、荷兰 (NL) 和中国 (CN) 队列中炎症性肠病 (IBD)、UC 和 CD 的 vOTU 与健康对照组的系数值。(f) 三种具有潜在致病基因的代表性噬菌体(即vOTU 89、90和689)的基因组结构。
05
UC 和 CD 之间有明显的真菌群特征
接下来,他们在宏基因组中研究了真菌和原生动物等真核生物物种(方法)。在日本 4D 队列中,与对照组相比,IBD、UC 和 CD 患者中仅分别鉴定出 2、3 和 1 种 FDR < 0.1 的真菌。由于 FDR < 0.1 的真核生物物种的检测率较低,因此使用P值进行探索性分析。
对日本 4D 队列中检测到的真核生物种类进行比较,发现 IBD、UC 和 CD 患者与对照组相比,分别有 6、3 和 2 种真菌发生显著变化。在 CD 中显著富集的真菌包括酿酒酵母和汉逊德巴约酵母,而在 UC 中富集的真菌包括奇异酵母(Saccharomyces paradoxus)和库德里阿兹威氏酵母(Saccharomyces kudriavzevii)。仅在 UC 中观察到Exophiala mesophila的显著减少(图 5a),没有一种归类为原生生物的物种与 IBD 有显著关联(图 5a)。
为了验证日本 4D 队列的结果,他们比较了外部数据集中的真核生物物种,发现酿酒酵母在美国 CD 患者中也富集和 ES 队列但在 CN 队列中 CD 显著减少。在日本 4D 队列中 UC 中富集的奇异酵母在美国队列的 UC 中也显著增加。在 ES 队列中,Saccharomyces paradoxus和Saccharomyces kudriavzevii在CD中显著富集。在 ES 队列中, Debayomyces hansenii在UC 中显著增加(图 5a)。
为了研究与疾病相关的微生物之间的相互作用网络,他们构建了日本 4D 队列中重要细菌、病毒和真菌物种之间的相关网络。为了消除疾病对相关性的混杂影响,他们评估了健康对照中的相关性(图 5b-c),发现显著不同于零的关联和重要的相互作用(例如,预期宿主和噬菌体、真菌和细菌以及病原体和细菌之间的联系)。
在UC相关物种的关联网络中,在UC中减少的产SCFA细菌如Blautia massiliensis、Dorea longicatena和未分类的Ruminococcus形成聚类(图5b)。此外,他们观察到几个噬菌体及其预测宿主的聚类(例如,vOTUs 160、161、162和164的Ruminococcus bromii和Ruminococcus噬菌体;vOTU289、291、293和294的Fusicatenibacter saccharivorans和Fusicatenibacter噬菌体)。在CD相关物种的关联网络中,产SCFA细菌如Eubacterium vntriosum、Blautia obeum和未分类的Clostridiales形成一个大聚类,其中还包括几个预测会感染它们的噬菌体。相反,在 CD 中显著增加的大肠杆菌和酿酒酵母与这些物种呈负相关。大肠杆菌噬菌体 (vOTU77) 与大肠杆菌显著相关(图 5c)。UC 和 CD 相关物种彼此之间具有很强的相关性并形成多个聚类,这一发现凸显了它们在患者肠道微生物组中相互作用的重要性。
为了验证在日本 4D 队列中构建的关联网络,他们分析了美国、西班牙、荷兰和中国队列中的网络。对 UC 相关物种的分析表明,在US和ES队列中,Ruminococcus噬菌体(vOTU160、161、162 和 164)也与Ruminococcus bromii显著相关,并且这种关联在 NL 队列中也观察到(图 5d)。同时,他们对 CD 相关物种的分析表明,vOTU77(大肠杆菌噬菌体)与CN 和 ES 队列中的大肠杆菌显著相关。Anaerostipes噬菌体(vOTU758)与Anaerostipes hadrus以及链球菌噬菌体(vOTU689)与链球菌属之间存在显著的正关联。在 ES 队列中也观察到了相关性。在US队列中,酿酒酵母与Eubacterium ventriosum 和Clostridiales sp.负相关。在所有队列中也观察到大肠杆菌与 SCFA 产生菌(Anaerostipes hadrus、Blautia obeum和Clostridiales sp.)之间的负相关(图 5e)。这些结果表明在日本 4D 队列中发现的相关性网络的稳健性。
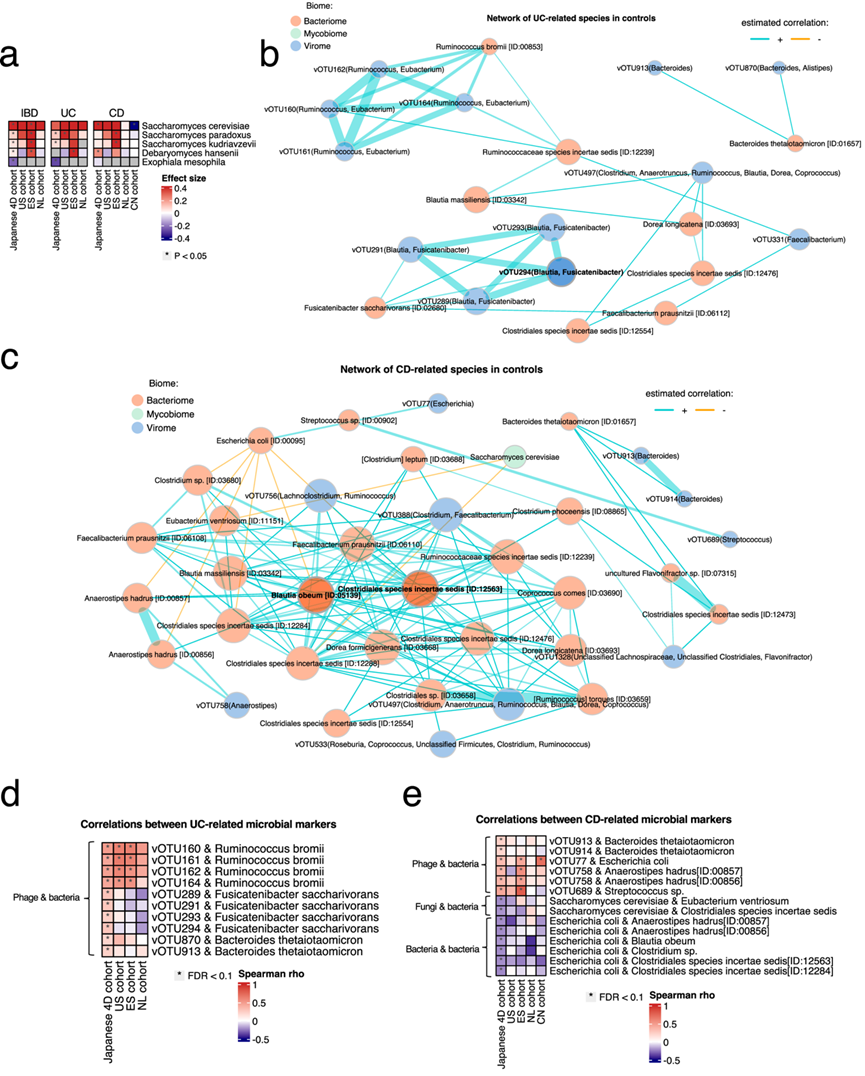
图5. UC 和 CD 中的真菌组特征和多真菌组相互作用。
(a) 热图描绘了日本 4D 队列、美国 (US)、西班牙 (ES)、荷兰 (NL) 和中国 (CN) 队列中炎症性肠病 (IBD)、溃疡性结肠炎 (UC) 和克罗恩病 (CD) 的真菌组与健康对照的系数值。(b) 网络图显示了健康对照中 UC 相关细菌组、病毒组和真菌组之间的界内和跨界相互作用。(c) 网络图显示了健康对照中 CD 相关细菌组、病毒组和真菌组之间的界内和跨界相互作用。(d-e) 该热图显示了评估日本 4D 队列中健康对照中UC 相关物种和CD 相关物种之间关联 的系数值。
+ + + + + + + + + + +
结 论
本项研究对日本 4D 队列中的 UC 和 CD 患者及健康对照者的粪便进行宏基因组测序,分析细菌分类群、基因功能以及抗菌基因、噬菌体和真菌。分析了来自美国、西班牙、荷兰和中国的外部宏基因组数据集,以验证多生物组结果。粪肠球菌和双歧杆菌在两种疾病中都富集。大肠杆菌富集是 CD 的特征,并且与参与外排泵的多种抗生素耐药性基因和粘附侵入性大肠杆菌毒力因子有关。病毒组的变化与细菌组的变化相关,包括编码致病基因的噬菌体丰度增加。奇异酵母菌和酿酒酵母分别在 UC 和 CD 中富集。酿酒酵母和大肠杆菌与 CD 中SCFA产生菌呈负相关。UC 和 CD 中的多生物群特征及其相互作用在日本和其他国家之间表现出高度相似性。由于细菌、噬菌体和真菌与 UC 和 CD 中的 SCFA 生产者和致病菌形成了多界内或跨界网络中心,因此针对相互作用网络的方法可能具有治疗前景。
+ + + + +



