

 English
English文献解读|Adv Sci(14.3):乳腺导管癌的蛋白质组学图谱揭示肿瘤进展特征和治疗靶点
✦ +
+
论文ID
原名:Proteogenomic Landscape of Breast Ductal Carcinoma Reveals Tumor Progression Characteristics and Therapeutic Targets
译名:乳腺导管癌的蛋白质组学图谱揭示肿瘤进展特征和治疗靶点
期刊:Advanced Science
影响因子:14.3
发表时间:2024.10.17
DOI号:10.1002/advs.202401041
背 景
乳腺癌 (BC) 是女性最常见的癌症之一,其形态、分子表达谱和临床病程具有高度异质性。乳腺导管癌 (BRDC) 是 BC 最常见的类型,具有独特的临床和病理特征。组织病理学上,人 BRDC 的经典进展模型是一个线性多步骤过程,始于导管增生 (DH),进展为导管原位癌 (DCIS),并演变为浸润性导管癌 (IDC)。根据组织病理学标准[包括激素受体(雌激素受体和/或孕激素受体)和/或人表皮生长因子受体 2 (HER2) 的表达],IDC 分为四种主要临床亚型:管腔 A、管腔 B、HER2 富集型和 TNBC。
实验设计
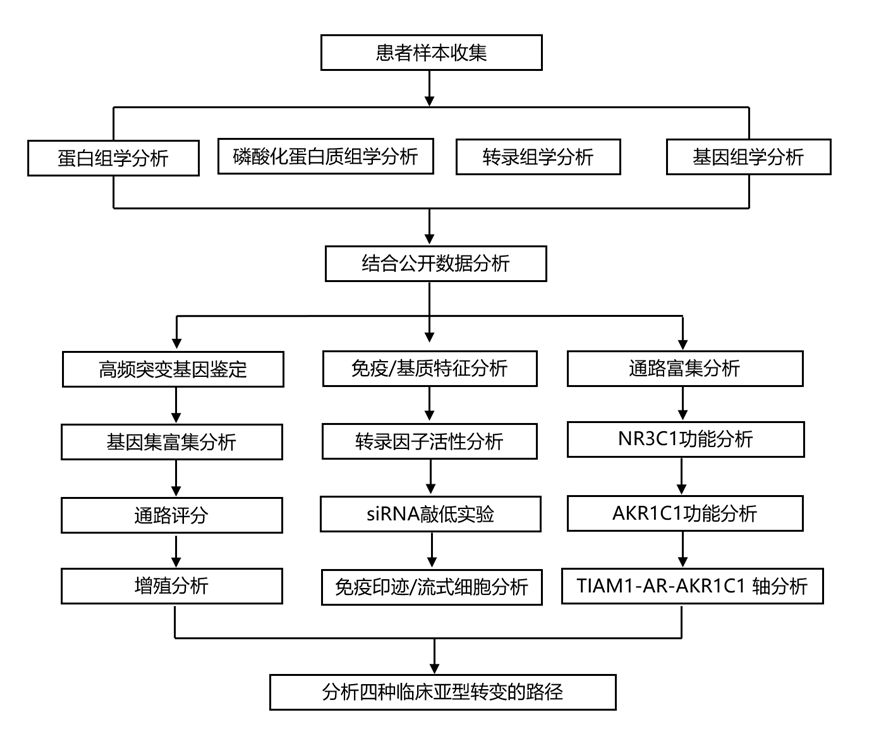
结 果
01

BRDC 进展的蛋白质组学分析
本研究共收集了 168 名患有恶性和良性乳腺疾病且未接受过治疗的女性患者的 224 个样本。随后,三位专家病理学家将本研究队列中的 224 个样本分为 4 个进展阶段,包括正常导管上皮组织(Normal)、导管增生(DH)、导管原位癌(DCIS)和浸润性导管癌(IDC)。DH 样本包括正常导管增生(UDH)和非典型导管增生(ADH)。
用基于质谱 (MS) 的非标记定量策略对 224 个样本进行了蛋白质组学分析,使用 Fe-NTA 富集策略对 49 个样本进行了磷酸化蛋白质组学分析,并对 79 个样本进行了全外显子组测序 (WES),以检测癌症基因组中任何可能的遗传变异。此外,还对 42 个样本进行了 转录组分析(RNA-seq),本研究在多组学水平上揭示了 BRDC 的进展特征(图1A)。
WES 数据导致平均靶标覆盖率为 131.1 倍,并鉴定出 14885 个遗传变异事件。他们鉴定了 18 个高频突变(TP53、PIK3CA、KMT2D、FAT3、ARID1A、GATA3、PREX2、APC、ERBB2、FAT1、CDH1、NOTCH2、PTEN、BRCA2、CIC、MAP3K1、AKT1和AKAP9)(图 1C)。值得注意的是,整个队列中TP53基因的突变频率为 34%,显著突变始于 DCIS 期(图 1C)。与Pareja的DCIS队列相比,本研究的DCIS队列中的高频突变与Pareja队列几乎一致(图 1D)。与Jiang的IDC队列相比,本研究的IDC队列中已知的BC高频突变与Jiang的IDC队列几乎一致(图 1E)。这些结果进一步证明本研究中的基因组测序数据的可靠性。
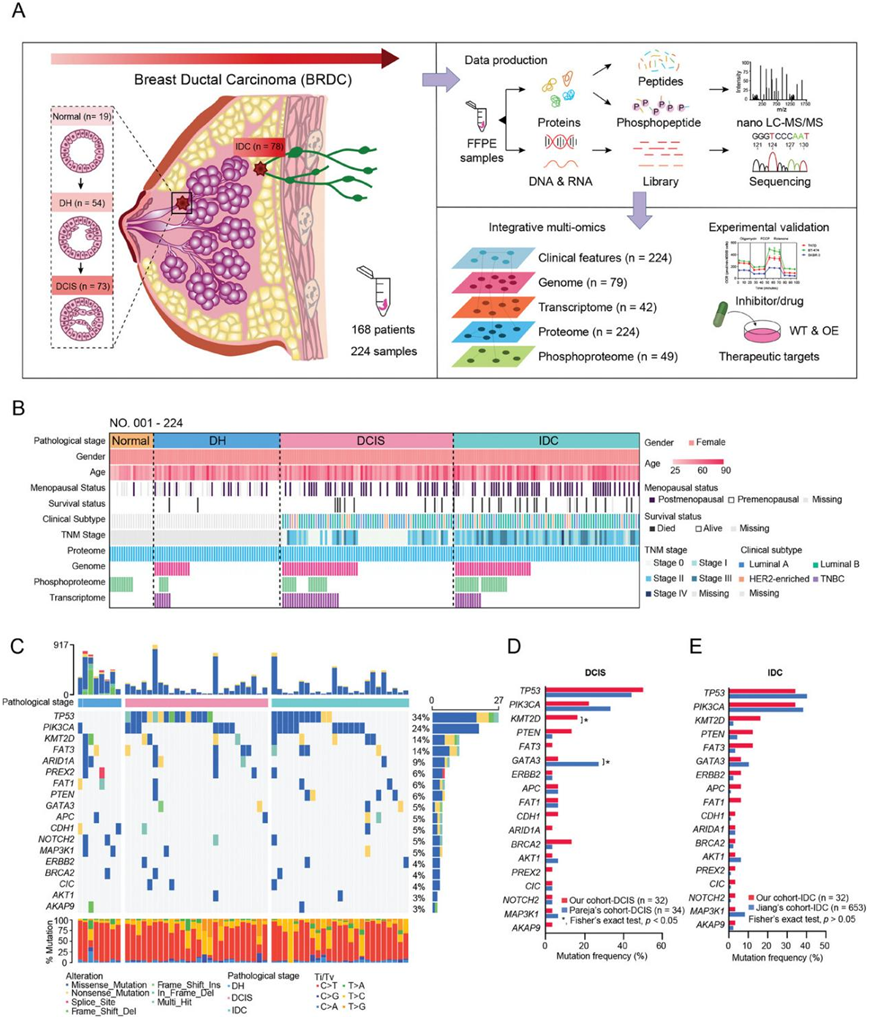
图1. BRDC 进展的蛋白质组学分析。
(A) 实验设计概述。(B) 热图中显示了研究队列的临床参数。(C) 所有 79 个 BRDC 进展样本的基因图谱和相关病理阶段。(D-E) 不同队列的比较。
02
TP53突变相关ESR1过表达参与BRDC的肿瘤发生
为了阐明DH进展为DCIS的第一步,他们研究了BRDC进展中的新突变,发现TP53突变是BRDC的高频事件,并且早在肿瘤发生阶段就发生了突变,在DCIS中发生的频率为50%,而在DH中没有观察到TP53突变(图 1C,图2A)。其次, TP53突变组TP53表达量显著高于TP53野生型组(图 2B)。一致地,DCIS中TP53的表达量显著高于DH(图 2B)。进一步的基因集富集分析(GSEA)显示,与TP53野生型组相比, TP53突变组的胆固醇稳态通路显著富集(图 2C)。此外,相关性分析结果显示,胆固醇稳态通路的富集评分与TP53的表达呈显著正相关(图 2D)。基于单样本基因集变异分析 (ssGSEA)的通路评分表明,与野生型TP53组和 DH 相比, TP53突变组和 DCIS中的胆固醇稳态通路显著上调(图 2E)。综上所述,这些结果表明突变型 TP53 可能参与调节胆固醇稳态。
先前的研究表明,多个基因可以特异性地与突变的TP53结合并参与调控恶性表型。进一步,他们收集了一组与突变型TP53形成复合物的蛋白质,其中固醇调节元件结合转录因子2 (SREBF2)的表达在TP53突变组中显著高于TP53野生型组(图 2F)。基于RNA-seq数据推断TP53突变组和DCIS中SREBF2的转录活性。结果表明,TP53突变组和DCIS中SREBF2的转录活性高于野生型TP53组和DH(图 2G)。GSEA显示胆固醇稳态通路在TP53突变组和DCIS中富集(图 2H)。从另一个角度,他们分析了特定转录因子(TF)与靶基因(TG)之间的关系,以及它们在BRDC进展过程中的动态变化。值得一提的是,DCIS的特异性TF,如固醇调节元件结合转录因子1 (SREBF1),SREBF2和雌激素受体1 (ESR1)主要参与类固醇代谢过程。与野生型TP53组和DH相比,胆固醇生物合成相关蛋白[羊毛甾醇合酶(LSS),固醇-C5-去饱和酶(SC5D)和甲羟戊酸二磷酸脱羧酶(MVD)]在TP53突变组和DCIS中在mRNA和蛋白质水平上均上调(图 2I-J)。这些结果表明突变型TP53与SREBF2共同参与DCIS中胆固醇稳态的调节。
细胞内胆固醇水平反映了胆固醇合成、运输和酯化之间的动态平衡。相关性分析结果显示,SREBF2与类固醇激素生物合成富集评分呈显著正相关,提示在DCIS中合成的胆固醇可能代谢为类固醇激素(图 2K)。在TP53突变组和DCIS 中雌激素合成相关蛋白上调(图2L)。这些结果提示,细胞内胆固醇可能通过StAR相关脂质转移结构域3 (STARD3)转运至线粒体,代谢为雌激素(图 2L)。在6种类固醇激素受体[雌激素受体1(ESR1)、雌激素受体2(ESR2)、雄激素受体(AR)、孕激素受体(PGR)、核受体亚家族3组C成员1(NR3C1)和核受体亚家族3组C成员2(NR3C2)中,仅TP53突变组和DCIS的ESR1活性和表达水平高于TP53野生型组和DH(图 2M)。为了进一步评估转录因子ESR1在DCIS中的作用,他们结合了来自CHEA转录因子靶标数据集,在TP53野生型组和DCIS中,细胞增殖相关通路均上调(图 2N)。TP53突变组和DCIS的多基因增殖评分(MGPS)显著高于TP53野生型组和DH。此外,他们通过免疫组织化学 (IHC) 检查了本研究队列中突变型 TP53 和 ESR1 的表达水平,证实 DCIS 中突变型 TP53 和 ESR1 的表达高于 DH(图2O)。
为了验证SREBF2是否参与胆固醇合成并进而调控ESR1的表达,他们利用针对SRBEF2的siRNA敲低MCF10DCIS.COM细胞中SRBEF2的表达,通过mRNA和蛋白表达水平验证 SRBEF2敲低细胞的有效性(图2P-Q)。接下来他们检测了胆固醇合成基因(LSS、SC5D和MVD)的转录水平。结果显示,与野生型MCF10DCIS.COM细胞相比, SRBEF2敲低的MCF10DCIS.COM细胞中胆固醇合成相关蛋白(LSS、SC5D和MVD)的转录水平显著下调(图 2Q)。为了验证SREBF2敲低对胆固醇及其下游雌激素合成水平的影响,他们通过ELISA检测了SRBEF2敲低的MCF10DCIS.COM细胞中胆固醇及其下游雌激素的合成水平。在SREBF2敲低后,MCF10DCIS.COM细胞中胆固醇及其下游雌激素的合成水平降低(图 2R-S)。这些结果提示,SREBF2参与了胆固醇及其下游雌激素的合成。据报道,雌激素可以增强ESR1的表达。与野生型MCF10DCIS.COM细胞相比,SRBEF2敲低的MCF10DCIS.COM细胞中ESR1的表达水平显著下调(图2T)。此外,与野生型MCF10DCIS.COM细胞相比,他们发现SRBEF2敲低的MCF10DCIS.COM细胞的细胞增殖受到明显抑制(图 2U),这些结果进一步证明SRBEF2的激活导致胆固醇合成基因增多,进而引起ESR1的上调,进而促进细胞增殖。综上所述,这些结果表明TP53突变增加 ESR1 活性和表达,进一步促进细胞增殖,导致致癌作用(图 2V)。
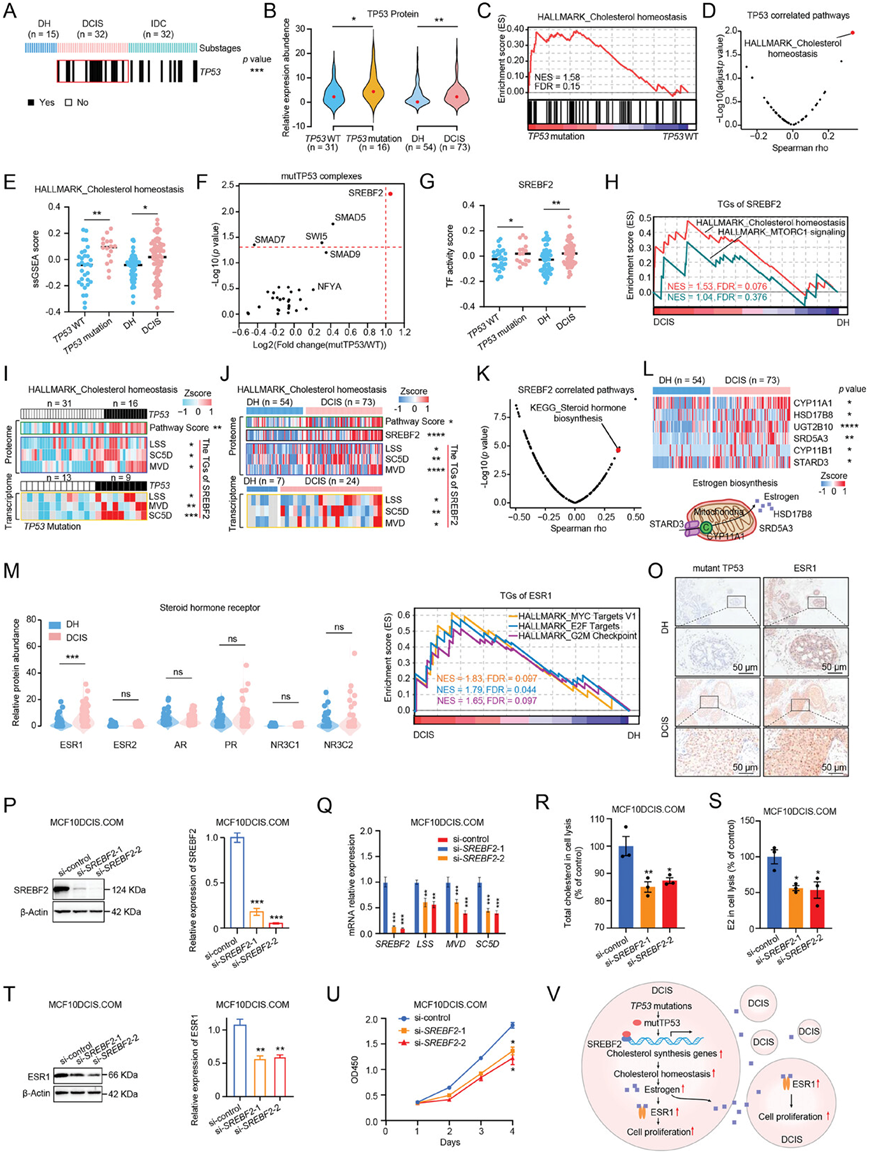
图2. TP53突变相关的 ESR1 过表达与 BRDC 的肿瘤发生有关。
(A) BRDC 进展不同阶段中的TP53突变。(B) 小提琴图显示TP53 蛋白表达。(C) 基因集富集分析 (GSEA)。(D) 火山图显示 TP53 表达水平与 ssGSEA 通路评分之间的相关性。(E) 胆固醇稳态通路评分比较。(F) 突变型TP53复合物的蛋白表达水平。(G) SREBF2 转录活性比较。(H) GSEA 显示 SREBF2 的 TG 参与了 DCIS 中的胆固醇稳态调控。(I) 热图显示 TP53 突变和 TP53 野生型组中胆固醇稳态的通路评分、SREBF2 的 TG 的蛋白质表达水平以及 SREBF2 的 TG 的 mRNA 水平。(J) 热图显示 DCIS 和 DH 中的胆固醇稳态通路得分、SREBF2 蛋白质表达水平、SREBF2 TG 蛋白质表达水平以及 SREBF2 TG mRNA 水平。(K) 火山图显示通路得分与 SREBF2 表达水平之间的相关性。(L) 热图显示 DH和 DCIS(上)中雌激素合成相关蛋白的蛋白质表达水平,线粒体中雌激素合成的示意图(下)。(M) 小提琴图显示 DH和 DCIS中固醇激素受体的蛋白质表达水平。(N) GSEA 显示 ESR1 的 TG 参与 DCIS 中的增殖相关通路。(O) DH 和 DCIS 组织上 TP53 和 ESR1 的代表性 IHC 图像。(P) 免疫印迹分析。(Q) 实时 PCR 分析。(R) SRBEF2敲低 MCF10DCIS.COM 细胞裂解中的胆固醇含量。(S) SRBEF2敲低 MCF10DCIS.COM 细胞裂解中的 E2 含量。(T) 免疫印迹分析。(U) 细胞增殖分析。(V) 描述TP53突变相关 ESR1 过表达的功能影响的简要模型。
03
6q21扩增相关的NR3C1过表达参与DCIS_Pure中肿瘤细胞的免疫逃逸
结合组织病理学信息,他们将队列中的 73 个 DCIS 样本分为两个亚组:DCIS_Pure(纯 DCIS)和 DCIS_adjIDC(毗邻 IDC 的 DCIS,侵袭性癌症的 DCIS 区域)。基于蛋白质组学数据研究了队列中的肿瘤微环境成分。值得注意的是,DCIS_Pure 显示出最低的免疫评分(图 3A)。在 DCIS_Pure 中,CD8+ Tem、CD8+ Tcm 和 CD8+ T 细胞的丰度处于最低水平,这与 CD8+ T 细胞标志物 CD8A 的表达水平一致(图 3B)。蛋白质组学和磷酸化蛋白质组学分析表明,与DH、DCIS和IDC相比,正常组中炎症反应相关通路显著上调(图 3B )。此外,他们发现DCIS_Pure中免疫相关通路下调,包括细胞因子-细胞因子受体相互作用、T细胞受体信号转导、B细胞受体信号转导和JAK-STAT信号转导(图 3B)。这些结果表明 DCIS_Pure 在 BRDC 进展中处于免疫静止阶段。
相反,类固醇激素受体NR3C1在mRNA和蛋白质水平上都达到峰值(图 3B-C)。据报道,NR3C1是炎症的重要下调因子。值得注意的是,NR3C1的表达与免疫评分之间存在显著的负相关性,这表明NR3C1可能参与DCIS_Pure中的免疫静止。NR3C1的总共5个TF(IRF3、PRDM1、SRF、TCF3和GATA6)在其蛋白质表达水平和拷贝数改变(CNA)之间显示出顺式效应(图 3D)。其中,他们在6q21上鉴定出一个PR-domain containing 1(PRDM1)的顺式效应,且其在DCIS_Pure中的蛋白表达水平高于DH(图 3D-E)。此外,他们发现NR3C1与PRDM1的蛋白表达之间存在显著的正相关性(图 3E)。
转录活性分析表明,核因子κB亚基1(NF κ B1)和核因子NF-κB P65亚基(RelA)在TNF- α信号通过NF-κB通路中的转录活性在DCIS_Pure中显著降低(图 3F)。NF κ B1和RelA复合物的转录活性评分与免疫评分和细胞因子-细胞因子受体相互作用通路的富集评分呈显著正相关性(图 3G)。细胞因子 CXC 基序趋化因子配体 12(CXCL12)和 C-X3-C 基序趋化因子配体 1(CX3CL1),NF κ B1 和 RelA 的 TG,在 DCIS_Pure 中的 mRNA 和蛋白质水平上均显著下调(图 3H)。先前的研究报告称,CXCL12 和 CX3CL1 可以影响 CD8+ T 细胞。在本研究的蛋白质组学数据中,CXCL12 和 CX3CL1 与 CD8 亚基 alpha (CD8A)显著正相关(图 3I),并且 CD8A 在 DCIS_Pure 中下调(图 3B)。
有趣的是,他们观察到DH和DCIS_adjIDC的免疫评分高于DCIS_Pure,提示DCIS_Pure中存在免疫逃逸,而DCIS_adjIDC中存在免疫激活(图 3A)。为了探究DCIS_adjIDC中免疫激活的原因,他们进行了顺式效应分析,发现PPARD是顺式效应基因,导致PPARD编码的蛋白质(过氧化物酶体增殖激活受体δ:PPARD)在DCIS_adjIDC中上调,转录因子PPARD参与免疫调控。
另一方面,IHC证明DCIS_Pure中CD8+ T细胞的丰度低于DH和DCIS_adjIDC,而NR3C1的表达水平在DCIS_Pure中高于DH和DCIS_adjIDC(图 3J)。综上所述,从DH到DCIS_Pure,6q21增益增加了PRDM1的表达并进一步上调NR3C1的表达,通过抑制NF κB介导的TNF- α信号通路导致CX3CL1和CXCL12的表达下调。CX3CL1和CXCL12的下调表达导致它们对CD8+ T细胞的趋化作用降低,从而导致DCIS_Pure中肿瘤细胞的免疫逃逸(图 3K)。
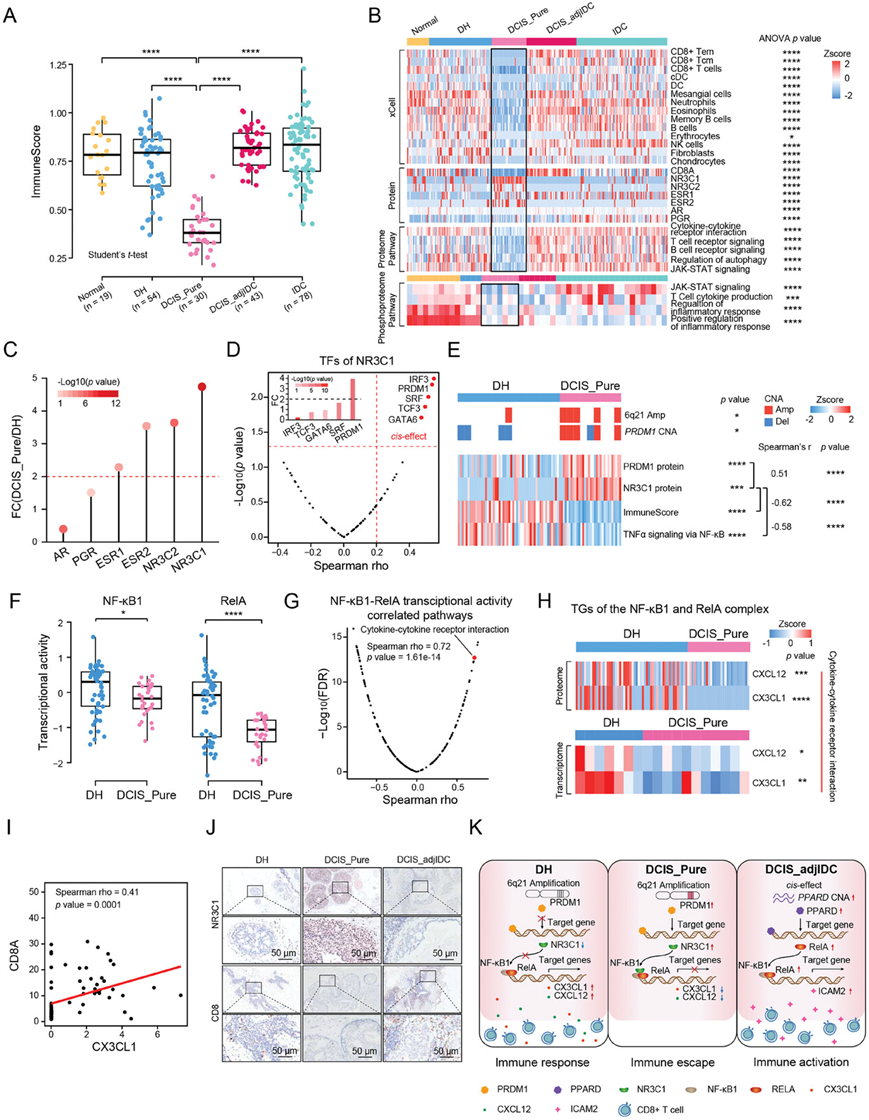
图3. 6q21 扩增相关的 NR3C1 过表达与 DCIS_Pure 中肿瘤细胞的免疫逃逸有关。
(A) 箱线图显示了 BRDC 进展不同阶段的免疫评分。(B) 热图显示了 BRDC 进展阶段中选定的单个基因/蛋白质和通路的细胞类型组成和活性。(C) 固醇激素受体蛋白质表达水平的比较。(D) 火山图显示 NR3C1 TF 的蛋白质表达水平与其相应 CNA 之间的相关性。(E) 热图显示DH 和 DCIS_Pure 中PRDM1的拷贝数变异 (CNA) 、PRDM1 和 NR3C1 的蛋白质丰度、免疫评分以及 TNF-α 通过 NF-ĸB 信号传导的通路评分。(F) 箱线图显示 DH和 DCIS_Pure中 NF-ĸB1 和 RelA 的转录活性。(G) 火山图显示 NF-ĸB1 和 RelA 的转录因子活性与 ssGSEA 通路评分之间的相关性。(H) 热图显示 DH 和 DCIS_Pure 中 CXCL12 和 CX3CL1 的蛋白质表达水平(上)以及 CXCL12 和 CX3CL1 的 mRNA 水平(下)。(I) CX3CL1 和 CD8A 蛋白质表达水平的 Spearman 等级相关性。(J) 代表性 IHC 图像。(K) DH、DCIS_Pure 和 DCIS_adjIDC 之间的免疫改变示意图。
04
NR3C1 过表达与肿瘤细胞的免疫逃逸有关
为了验证PRDM1是否调控NR3C1的表达,他们利用针对PRDM1的siRNA敲低MCF10DCIS.COM细胞中PRDM1的表达,通过mRNA和蛋白表达水平验证PRDM1敲低细胞的有效性(图 4A-B)。为了验证PRDM1是否调控NR3C1的表达,他们检测了NR3C1的表达水平,发现PRDM1敲低后NR3C1的表达水平显著降低(图 4B),这些结果说明PRDM1敲低降低了NR3C1的表达水平。接下来,为了验证PRDM1敲低对CX3CL1和CXCL12水平表达的影响,他们检测了CX3CL1和CXCL12的转录水平和蛋白表达水平。上述结果显示,与野生型MCF10DCIS.COM细胞相比,PRDM1敲低的MCF10DCIS.COM细胞中CX3CL1和CXCL12的转录水平和蛋白表达均显著上调(图 4C-F)。此外,他们检测了PRDM1敲低的MCF10DCIS.COM细胞中CX3CL1和CXCL12的释放水平,发现PRDM1敲低后,培养基(CM)中CX3CL1和CXCL12的表达水平增加(图 4G-H)。
为了验证NR3C1通过抑制NF-κB介导的TNF-α信号通路来调控CX3CL1和CXCL12的下调表达,他们利用针对NR3C1的siRNA来敲低MCF10DCIS.COM细胞中NR3C1的表达。在mRNA和蛋白质水平上验证了 NR3C1敲低细胞的效果(图4I-J)。此外,在用NF-κB抑制剂BAY 11-7082 (10 µm)处理后,NF-κB信号通路发生下调,如p65磷酸化水平所示(图 4K )。接下来,他们检测了CX3CL1和CXCL12的转录水平和蛋白质表达水平。上述结果显示,与野生型MCF10DCIS.COM细胞相比,NR3C1敲低的MCF10DCIS.COM细胞中CX3CL1和CXCL12的转录水平和蛋白表达水平显著上调(图 4L-O)。然而,用BAY 11-7082 (10 µm)处理NR3C1敲低的MCF10DCIS.COM细胞后,CX3CL1和CXCL12的转录水平和蛋白表达水平显著下调,这表明NR3C1敲低引起的CX3CL1和CXCL12的上调趋势由BAY 11-7082抑制(图 4L-O)。此外,他们检测到了NR3C1敲低的MCF10DCIS.COM细胞中CX3CL1和CXCL12的释放水平。在NR3C1敲低后CM中CX3CL1和CXCL12的表达水平增加(图4P-Q)。然而,在用BAY 11-7082 (10 µm) 处理NR3C1敲低的MCF10DCIS.COM细胞后的CM中,其CX3CL1和CXCL12的释放水平降低(图 4P-Q)。这些结果进一步证实NR3C1通过抑制NF-κB介导的TNF-α信号通路来下调CX3CL1和CXCL12的表达水平。
此外,之前研究还报道 CXCL12 和 CX3CL1 能够趋化 CD8+ T 细胞。在本研究的蛋白质组学数据中,CXCL12 和 CX3CL1与CD8A 呈显著正相关,这表明 CXCL12 和 CX3CL1 可能具有趋化 CD8+ T 细胞的能力。为了进一步验证上述假设,他们从 BC 患者的外周血单个核细胞(PBMC)中分离 CD8+ T 细胞。通过流式细胞技术检测分离的 CD8+ T 细胞的纯度(图 4R)。然后,他们进行了双室 CD8+ T 细胞迁移试验。与野生型 MCF10DCIS.COM 细胞的 CM 相比,NR3C1敲低的 MCF10DCIS.COM 细胞的CM 存在更多的 CD8+ T 细胞(图 4S)。这些结果进一步证实了CX3CL1和CXCL12的表达下调导致其对CD8 + T细胞的趋化性降低,从而导致DCIS_Pure中肿瘤细胞的免疫逃逸。
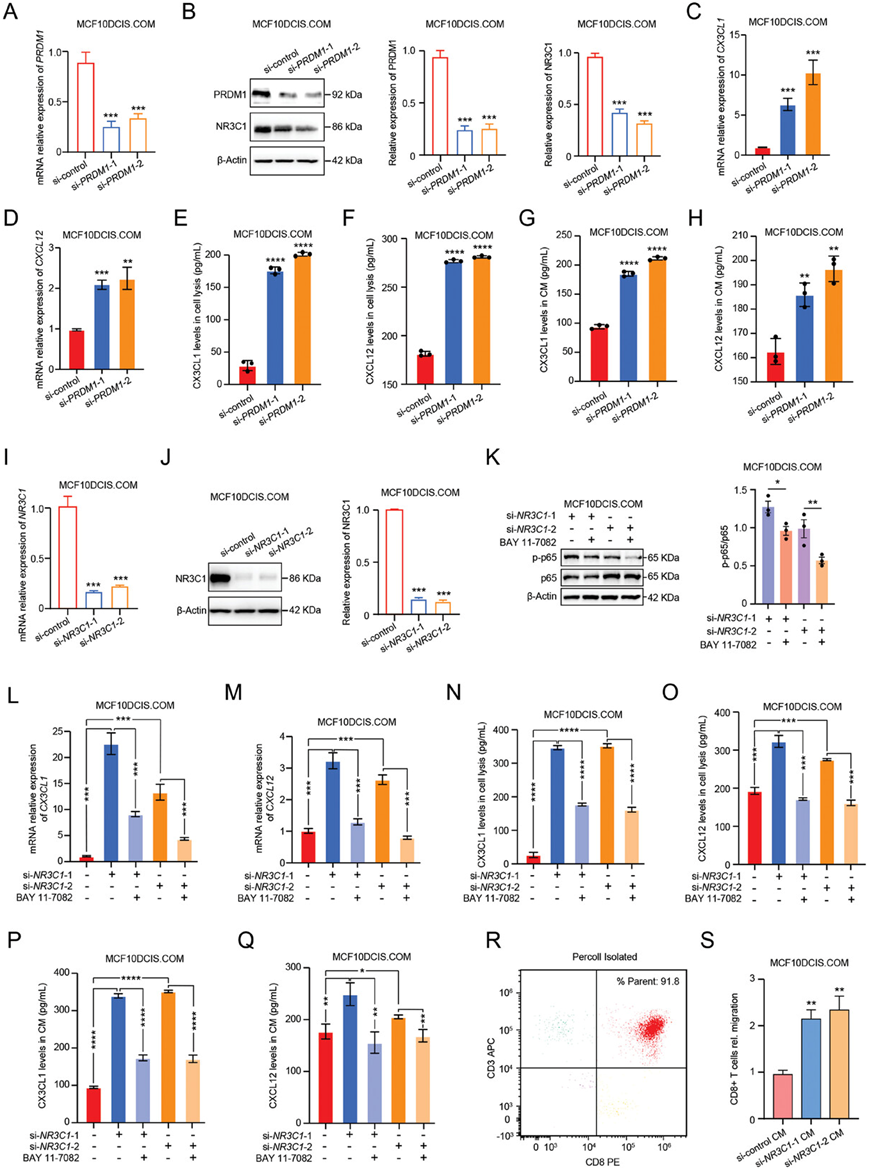
图4. NR3C1 过表达与肿瘤细胞的免疫逃逸有关。
(A) PRDM1敲低 MCF10DCIS.COM 细胞中PRDM1的实时 PCR 分析。(B)免疫印迹分析。(C-D) PRDM1敲低 MCF10DCIS.COM 细胞中CX3CL1和CXCL12的实时 PCR 分析。(E-F) ELISA 分析PRDM1敲低 MCF10DCIS.COM 细胞的细胞裂解物上清液中 CX3CL1 和 CXCL12 的蛋白质水平。(G-H) ELISA 分析PRDM1敲低 MCF10DCIS.COM 细胞的 CM 中 CX3CL1 和 CXCL12 的蛋白质水平。(I) 实时 PCR 分析PRDM1敲低 MCF10DCIS.COM 细胞中的NR3C1。(J-K) 代表性蛋白质印迹分析。(L-M) NR3C1 敲低 MCF10DCIS.COM 细胞或用 10 µ m BAY 11–7082处理的CX3CL1和CXCL12的实时PCR分析。(N-O) ELISA 分析NR3C1敲低 MCF10DCIS.COM 细胞或用 10 µm BAY 11–7082 处理的细胞裂解物上清液中 CX3CL1 和 CXCL12 的蛋白质水平。(P-Q) CX3CL1 和 CXCL12 的蛋白质水平。(R) 通过流式细胞技术分析从 BC 患者 PBMCs 中分离的CD8+ T 细胞的纯度。(S) NR3C1敲低 MCF10DCIS.COM 细胞的 CM 募集的 CD8+ T 细胞在 12 小时内进行双室迁移。
05
蛋白质组学分析表明TIAM1-AR-AKR1C1轴促进DCIS_adjIDC细胞侵袭和迁移
为了进一步研究DCIS_adjIDC如何具备侵袭能力并发展为IDC,他们筛选了DCIS_Pure与DCIS_adjIDC之间的差异表达分子(图 5A)。差异表达分子的通路富集分析显示,DCIS_adjIDC中富集的分子主要参与类固醇激素生物合成、O-聚糖生物合成、RHO GTP酶循环、HIF-1信号通路;DCIS_Pure富集的分子主要参与缬氨酸、亮氨酸、异亮氨酸降解、磷酸五糖、补体和凝血级联、黏着斑的调节(图 5A)。
为了确定 DCIS_Pure 和 DCIS_adjIDC 中基因组驱动因素的差异,他们比较了它们之间的基因组变异差异。多个染色体增益事件(例如染色体 21q22.11 和 4p16.1增益)在 DCIS_adjIDC中比在 DCIS_Pure中更频繁(图 5A)。21q22.11 中的 T 细胞淋巴瘤侵袭和转移 1 (TIAM1),具有显著正向顺式效应,在 DCIS_adjIDC 中上调(图 5A)。此外,在 CPTAC BC 队列中,TIAM1 在 mRNA 和蛋白质水平上也分别观察到显著正向顺式效应。TIAM1编码一种大蛋白,可作为 Rho GTPases 或 Rho 样 GTPases 的 GDP 到 GTP 交换因子,例如 RAC 家族小 GTPase 1 (RAC1)、RAC 家族小 GTPase 2 (RAC2)、RAC 家族小 GTPase 3 (RAC3)、RAS 同源物家族成员 A (RHOA) 和细胞分裂周期 42 (CDC42)。其中,RAC 家族成员在 DCIS_adjIDC 中上调,而 RHOA 和 CDC42 在 DCIS_Pure 中上调(图 5B)。TIAM1 的表达水平与 RAC3 的表达水平呈显著正相关性,但与 RAC1和 RAC2无关(图 5B-C)。
值得注意的是,RAC3的表达水平与类固醇激素生物合成通路的评分呈显著正相关(图 5D),并且类固醇激素生物合成途径在DCIS_adjIDC中上调。相应地,与DCIS_Pure相比,在6种类固醇激素受体中,只有AR的蛋白表达水平在DCIS_adjIDC中上调(图 5E),并且DCIS_adjIDC中AR的转录活性高于DCIS_Pure。此外,据报道,LIM激酶1 (LIMK1) 是Rho GTPases的特征下游效应物,并参与AR核易位。进一步研究发现,RAC3与LIMK1表达相关性为0.57,LIMK1与AR表达相关性为0.59(图 5F)。这些结果提示,激活的RAC3可能通过LIMK1促进AR的核转位。
他们进一步筛选了AR的靶基因,发现醛酮还原酶家族1成员C1 (AKR1C1) 在DCIS_adjIDC中mRNA和蛋白质水平均显著上调(图 5G)。据报道,AKR1C1参与调控活性氧(ROS)代谢过程。随后他们发现AKR1C1的表达水平与ROS通路评分呈显著负相关,在DCIS_adjIDC中ROS通路评分显著降低(图 5H),提示AKR1C1可以清除DCIS_adjIDC细胞中过量的ROS(图 5H)。清除细胞内过多的ROS可阻止肿瘤细胞凋亡,促进肿瘤细胞迁移和侵袭。令人惊讶的是,与DCIS_Pure相比,DCIS_adjIDC中的凋亡通路显著下调(图 5I-J)。此外,在人乳腺癌细胞HCC1806中敲低AKR1C1后,凋亡信号通路显著上调(图 5K-L),ROS的平均荧光强度(MFI)显著增加(图 5M)。另一方面,这与以前的报道一致,即ROS可能通过诱导缺氧诱导因子1α (HIF-1α) 诱导DCIS_adjIDC细胞侵袭和迁移,导致血管内皮生长因子 (VEGF) 上调,并抑制组织金属蛋白酶抑制剂1 (TIMP1),导致基质金属肽酶 (MMP) 上调(图 5N-O)。此外,IHC结果显示DCIS_adjIDC中TIAM1、AR和AKR1C1的表达高于DCIS_Pure(图 5P)。总体而言,这些结果表明 TIAM1-AR-AKR1C1 轴促进 DCIS_adjIDC 中的细胞侵袭和迁移(图5Q)。
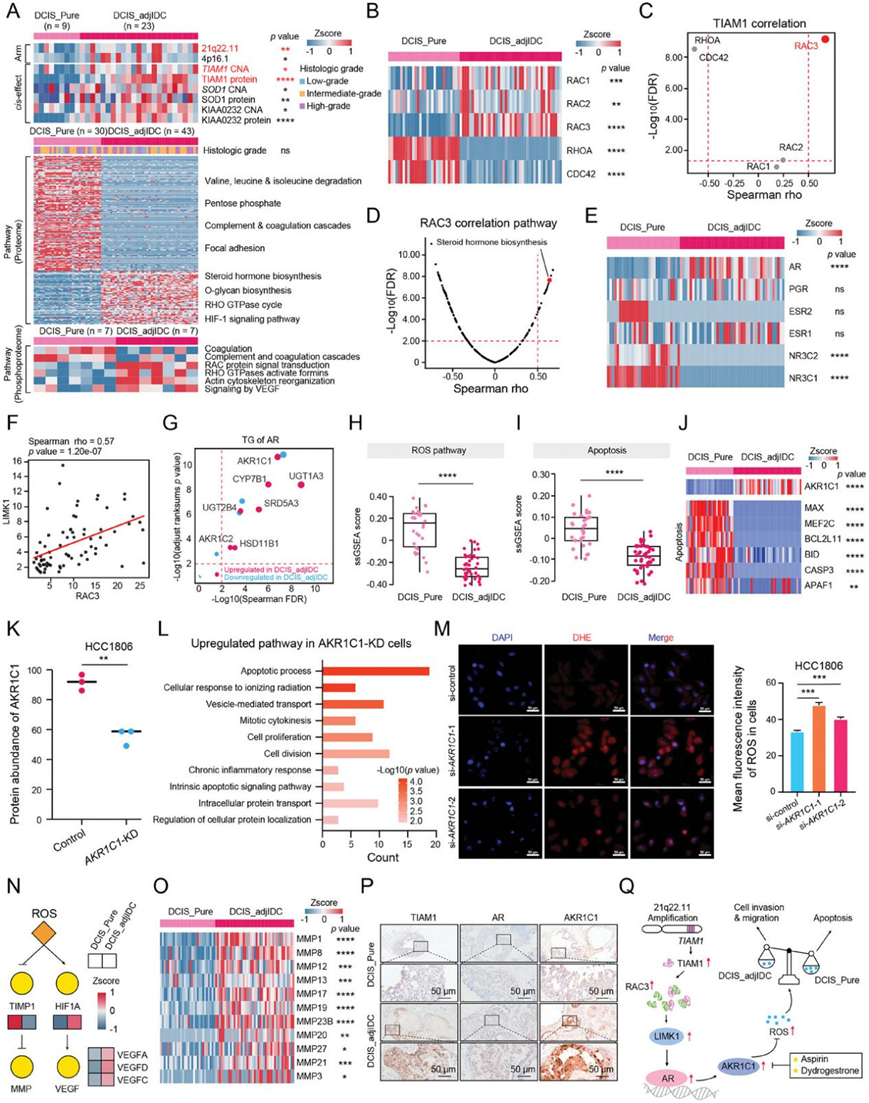
图5. 蛋白质组学分析表明 TIAM1-AR-AKR1C1 轴促进 DCIS_adjIDC 中的细胞侵袭和迁移。
(A) 热图显示 DCIS_Pure 和 DCIS_adjIDC 之间的多组学改变。(B) 热图显示 DCIS_Pure和 DCIS_adjIDC之间 Rho 样 GTPases 的蛋白质表达。(C) 鸟苷酸交换因子 TIAM1 与 Rho 样 GTPases 之间的 Spearman 相关系数。(D) 火山图显示 RAC3 表达水平与 ssGSEA 通路评分之间的相关性。(E) 类固醇激素受体的蛋白质表达水平。(F) RAC3 及其下游 LIMK1 蛋白质表达水平的 Spearman 等级相关性。(G) AR 的 TG。(H) ROS 通路评分比较。(I) 凋亡通路评分比较。(J) 凋亡相关蛋白的表达水平。(K) AKR1C1 的蛋白表达水平分析。(L) 敲低AKR1C1后,上调的通路。(M) 敲低AKR1C1后,ROS水平的DHE染色(左)和定量分析(右)。(N) ROS对侵袭和转移的调控。(O) 热图显示 DCIS_Pure和 DCIS_adjIDC中 MMP 的表达水平。(P) DCIS_Pure 和 DCIS_adjIDC 组织中 TIAM1、AR 和 AKR1C1 的代表性 IHC 图像。(Q) 该图表显示 DCIS_adjIDC 中的 TIAM1-AR-AKR1C1 改变及其对细胞侵袭和迁移的影响。
06
AKR1C1 是 BC 中潜在的可靶向蛋白
为了系统地验证TIAM1-AR-AKR1C1轴在BC中的作用和机制,他们进行了一系列实验研究。首先,为了验证TIAM1对细胞迁移和凋亡的影响,他们利用针对TIAM1的siRNA来敲低HCC1806中TIAM1的表达。通过mRNA和蛋白质表达水平验证TIAM1敲低细胞的有效性(图 6A)。接下来,他们对HCC1806或TIAM1敲低的细胞进行了细胞凋亡和迁移实验。与用 siRNA 处理的 HCC1806 相比,TIAM1敲低显著促进了细胞凋亡(图6B)。此外,他们通过 CCK-8 法检测了 TIAM1 敲低是否影响乳腺癌细胞 HCC1806 的生长。TIAM1敲低抑制了 HCC1806 细胞的增殖(图 6C)。此外,通过伤口愈合实验检测了HCC1806 或TIAM1敲低后的细胞的迁移能力。与 siRNA 处理的 HCC1806 相比, TIAM1敲低减少了细胞的迁移距离(图 6D)。综上所述,TIAM1 促进肿瘤细胞的迁移并抑制细胞凋亡。
进一步为了研究TIAM1是否通过AR核转位-AKR1C1 TF-TG对来影响细胞迁移和凋亡,他们提取了TIAM1敲低的HCC1806细胞的核蛋白,并检测了核蛋白AR的表达水平。TIAM1敲低后,细胞核中AR的表达水平显著降低(图 6E)。此外,他们进行免疫荧光观察发现在野生型HCC1806细胞中AR均匀分布于细胞核和细胞质中(图 6F),然而在TIAM1敲低的HCC1806细胞中AR与细胞核之间几乎没有共定位(图 6F)。与野生型HCC1806细胞相比,TIAM1敲低的HCC1086细胞中的转录水平显著降低(图6G)。这些结果表明TIAM1敲低抑制了 AR 核转位,并降低了 AKR1C1 的转录水平。然后,为了验证 AR 核转位在 AKR1C1 表达和 BC 进展中的作用,他们用 AR 核转位激活剂 5α-双氢睾酮 (5α-DHT) 处理 HCC1806 细胞。为了证明 5α-DHT 对 AR 核转位的影响,他们进行免疫荧光检查并观察到 5α-DHT(0.1 µm和1 µm)处理后 HCC1806 细胞中 AR 与细胞核之间更多的共定位。这些结果表明 5α-DHT 促进了 HCC1806 细胞中的 AR 核转位。然后,他们进行了细胞迁移试验并检测了 5α-DHT 处理后 AKR1C1 的表达水平。结果表明0.1μm和1μm 5α-DHT均可显著提高AKR1C1在mRNA和蛋白质水平的表达水平(图 6H-I)。此外,伤口愈合实验进一步显示0.1μm和1μm 5α-DHT均促进HCC1806细胞的迁移(图 6J)。综合以上结果,提示TIAM1促进AR核转位,进而调控AKR1C1的转录,可能引起细胞迁移。
接下来,为了确认 AKR1C1 在 BC 中的作用,他们选择了两种 AKR1C1 表达差异的人类 BC 细胞系 HCC1806 和 HCC1937 进行进一步研究(图 6K-L)。值得注意的是,过表达 AKR1C1 的细胞的迁移能力提高了(图 6M)。相反,使用针对AKR1C1的 siRNA来敲低 HCC1806 中 AKR1C1 的表达,通过蛋白表达情况验证AKR1C1敲低的效果(图 6N)。为了证明AKR1C1对细胞凋亡的影响,他们通过免疫印迹法检测了凋亡相关分子的表达。与siRNA处理的细胞相比,AKR1C1敲低的HCC1806细胞中凋亡相关分子caspase 3和caspase 8的表达水平显著升高(图 6N)。结果提示AKR1C1可以抑制细胞凋亡。此外,为了证明AKR1C1对细胞迁移的影响,通过伤口愈合和transwell实验检测了AKR1C1缺陷后HCC1806的细胞迁移能力。敲低AKR1C1显著减少了细胞迁移距离(图 6O)。这些结果表明 AKR1C1 促进细胞迁移。他们通过在TIAM1敲低的 HCC1806 细胞中过表达 AKR1C1 进行了挽救实验(图 6P)。值得注意的是,在TIAM1敲低的 HCC1806 细胞中过表达 AKR1C1 可有效逆转TIAM1沉默对细胞凋亡和迁移的影响(图6Q-R)。以上结果提示TIAM1促进AKR1C1的表达,从而加速HCC1806细胞的迁移并抑制其凋亡。
此外,基于 TIAM1-AR-AKR1C1 轴在乳腺癌进展中的作用,他们确定了靶向 AKR1C1 在体外治疗乳腺癌的潜在治疗效果。众所周知的水杨酸类药物阿司匹林可以抑制 AKR1C1 的活性。为了验证阿司匹林靶向 AKR1C1 对乳腺癌的潜在治疗作用,他们用最低有效剂量阿司匹林(0.5 mM)处理 HCC1806 细胞(图 6S)。结果表明,阿司匹林可以抑制细胞生长,并减少 HCC1806 的迁移距离(图 6T)。他们进一步采用了一种更有效的针对 AKR1C1 的 BC 抑制剂地屈孕酮来处理 HCC1806。结果显示,最低有效剂量的地屈孕酮 (10 µm) 显著抑制了 HCC1806 的细胞生长和细胞迁移(图 6U-V)。此外,10 µm 地屈孕酮对AKR1C1过表达的 HCC1937 细胞生长的抑制作用比野生型 HCC1937 更明显(图6W-X)。总之,这些结果揭示了 AKR1C1 在促进肿瘤细胞迁移和抑制细胞凋亡方面的作用,并且这种作用可以通过抑制剂来缓解。

图6. AKR1C1 是 BC 中潜在的靶标。
(A)免疫印迹分析。(B) 使用 Annexin V-PI 染色进行流式细胞分析,以评估TIAM1敲低的 HCC1806 细胞中的凋亡细胞百分比。(C) 记录TIAM1敲低后每 24 小时的 450 nm (OD450) 光密度,并据此绘制生长曲线。(D) 伤口愈合试验。(E) 免疫印迹分析。(F) 免疫荧光。(G-H) AKR1C1的实时 PCR 分析。(I) AKR1C1 的代表性免疫印迹分析。(J) 伤口愈合试验。(K-L) 免疫印迹分析。(M) 伤口愈合试验。 (N) 免疫印迹分析。(O) 伤口愈合试验。(P) 免疫印迹分析。(Q-R) 使用 Annexin V-PI 染色进行流式细胞分析以评估凋亡细胞的百分比。(S) 阿司匹林能够以剂量依赖性方式抑制 HCC1806 的细胞活力。(T) 生长曲线分析。(U) 地屈孕酮能够以剂量依赖性方式抑制 HCC1806 细胞活力。(V-X) 生长曲线分析。
07
DCIS 向 IDC 四种临床亚型转变的进展途径
目前,乳腺癌的多组学研究主要集中在IDC分期。根据ER、PR、HER2和(增殖标志蛋白KI67)KI67的表达情况,IDC分为四种主要临床亚型:管腔A、管腔B、HER2富集型和TNBC。为了说明四种IDC临床亚型的蛋白质组学特点,他们比较了四种IDC临床亚型之间的差异,分别鉴定了在管腔 A、管腔 B、HER2 富集和 TNBC 样本中显著过表达的 52、64、157 和 132 种蛋白质(图 7A)。使用 ConsensusPathDB 和 DAVID 数据库对富集蛋白进行聚类和聚类特异性富集分析,显示管腔 A、管腔 B、HER2 富集和 TNBC 样本中所代表的不同生物学过程和通路(图 7A)。具体而言,IDC-管腔 A 以能量代谢改变为特征。 IDC-管腔B 的特点是雌激素信号传导(即 ESR1 和 PR)和 mTOR 信号传导改变。IDC-HER2富集亚型与粘着斑和葡萄糖稳态相关。TNBC 与细胞周期、VEGF 信号通路、、TNF 信号传导和免疫相关通路改变。
为了阐明DCIS如何进展为不同亚型的IDC,他们通过GSEA对四种临床亚型的IDC和配对的DCIS_adjIDC样本进行了成对比较。DCIS_adjIDC样本相应地分为pre-管腔A、pre-管腔B、pre-HER2富集和pre-TNBC。GSEA 显示,与pre-管腔 A 相比,雌激素反应(例如 FHL2、AQP3、GFRA1、PODXL 等)、氧化磷酸化 (OXPHOS)(例如 ATP5PO、NDUFS7、COX6C、NDUFB1、ACAA1 等)和 TCA 循环(例如 IDH3B、DLST、SUCLA2 和 IDH3G)在 IDC-管腔 A 中上调(图7B)。与pre-管腔B相比,IDC-管腔B中的雌激素反应(例如PGR、SLC1A4、MUC1、WFS1、AQP3等)、OXPHOS(例如NDUFS7、COX6C、IDH3B、SUCLG1、ACAT1等)和脂肪酸代谢(例如SUCLG1、CA4、ADH7、CBR1、GRHPR等)均上调。与pre-HER2富集相比,IDC-HER2富集组中糖酵解(如PKM、CHPF2、ME2、SPAG4、STMN1等)和缺氧(如BHLHE40、SERPINE1、EXT1、SLC37A4、KIF5A等)上调。与TNBC前相比,IDC-TNBC中IL2-STAT5信号(如BCL2L1、TLR7、MUC1、PTH1R、CD48等)上调(图 7B-C)。这些数据提示,在DCIS向IDC的转变过程中,IDC-管腔 A定义为“需氧及TCA型”,IDC-管腔B定义为“需氧及脂肪酸代谢型”,IDC-HER2富集亚型定义为“缺氧及糖酵解型”,IDC-TNBC定义为“免疫型”。
为了验证从DCIS到IDC临床亚型的进展路径,他们选择了四种人乳腺癌细胞系[包括T47D(IDC-管腔A,HR+/HER2-)、BT-474(IDC-管腔B,HR+/HER2+)、SKBR-3(IDC-HER2-富集,HR-/HER2+)和MDA-MB-231(IDC-TNBC,HR-/HER2-),以更好地模拟在蛋白质组学数据中鉴定的IDC亚型模式。通过分析氧消耗率(OCR)和细胞外酸化率(ECAR),他们证实了两种管腔亚型细胞系T47D和BT-474比SKBR-3表现出更强的有氧活性,而SKBR-3表现出更强的糖酵解活性(图 7D-E),证明IDC-管腔亚型为“需氧型”,IDC-HER2富集型为“糖酵解型”的特点。
蛋白质组学数据显示,与pre-管腔亚型相比,OXPHOS和雌激素反应途径在IDC-管腔亚型中上调(图 7C)。为了靶向OXPHOS通路,IACS-010759作为临床级线粒体OXPHOS复合物I的小分子抑制剂,用于处理乳腺癌细胞。IACS-010759 (1μm)对T47D和BT-474细胞的抑制效果优于SKBR-3细胞(图 7F)。IACS-010759和雌激素受体抑制剂他莫昔芬联合使用,对T47D细胞的细胞活力的抑制效果优于单独使用他莫昔芬(图 7G)。此外,与pre-管腔A相比,IDC-管腔A中TCA循环通路上调(图 7C)。前期研究表明,TCA循环通路中的酰基辅酶A合成酶短链家族成员2(ACSS2)可能在低氧和缺脂条件下促进癌细胞生长。他们证实了ACSS2在T47D细胞中有高表达(图 7H)。用siRNA敲低ACSS2可抑制T47D细胞生长,而使用IACS-010759 (1μm)对siACSS2细胞的抑制效果更显著(图 7I)。
蛋白质组学数据显示,与pre-管腔B相比,IDC-管腔B中的脂肪酸氧化活性相对较高(图 7C)。正如预期的那样,在含有13C标记棕榈酸的培养基中培养的BT-474细胞中13C标记的乙酰辅酶A的形成最高(图 7J),这表明脂肪酸氧化活性在BT-474细胞中更高。为了靶向脂肪酸氧化通路,他们将etomixir (50µm)用作肉碱棕榈酰转移酶-1(CPT-1,脂肪酸氧化限速酶)抑制剂来处理乳腺癌细胞,它抑制了BT-474细胞的生长,但没有抑制SKBR-3细胞的生长(图 7K)。拉帕替尼作为HER2的小分子抑制剂,已用于治疗HER阳性乳腺癌患者。此外,他们发现Etomixir (50 µm)和拉帕替尼联合使用比单独使用拉帕替尼显著降低了 BT-474 细胞的细胞活力(图 7L)。
蛋白质组学数据显示,与pre-HER2富集相比,IDC-HER2富集的缺氧通路上调(图 7C)。针对缺氧通路,使用HIF-1α抑制剂px-478 (20 µm)处理乳腺癌细胞,抑制了SKBR-3细胞数量,但没有抑制管腔亚型细胞系(图 7M)。px-478 (20 µm)和拉帕替尼联合使用对SKBR-3细胞的抑制效果优于单独使用拉帕替尼(图 7N)。
蛋白质组学数据显示,与 pre-TNBC 亚型相比,IL2-STAT5 信号在 IDC-TNBC 中上调。为了研究针对 TNBC 乳腺癌细胞的 IL-2-STAT5 通路的潜力,他们用 IL-2 抑制剂 IL-2-IN-1 处理 MDA-MB-231 细胞。用 20 µm IL-2-IN-1 处理的 MDA-MB-231 细胞的生长受到显著抑制(图 7O)。这些结果表明 IL-2-IN-1 有可能成为抑制 TNBC 乳腺癌细胞的治疗剂。
综上所述,他们发现 IACS-010759 对 T47D 细胞的抑制作用优于对 BT-474 细胞和 SKBR-3 细胞的抑制作用。IACS-010759 作为管腔 A 型乳腺癌细胞的潜在治疗剂,与他莫昔芬联合使用对这些细胞表现出更好的抑制作用。此外,Etomixir 对 BT-474 细胞的抑制作用优于对 T47D 细胞和 SKBR-3 细胞的抑制作用。Etomixir 显示出作为抑制管腔 B 型乳腺癌细胞的治疗剂的潜力,与拉帕替尼联合使用可产生更好的抑制效果。px-478 显示出作为抑制 HER2 富集乳腺癌细胞的治疗剂的前景,与拉帕替尼联合使用在这方面更有效。最后,IL-2-IN-1 具有抑制 TNBC 细胞的潜力(图 7P)。
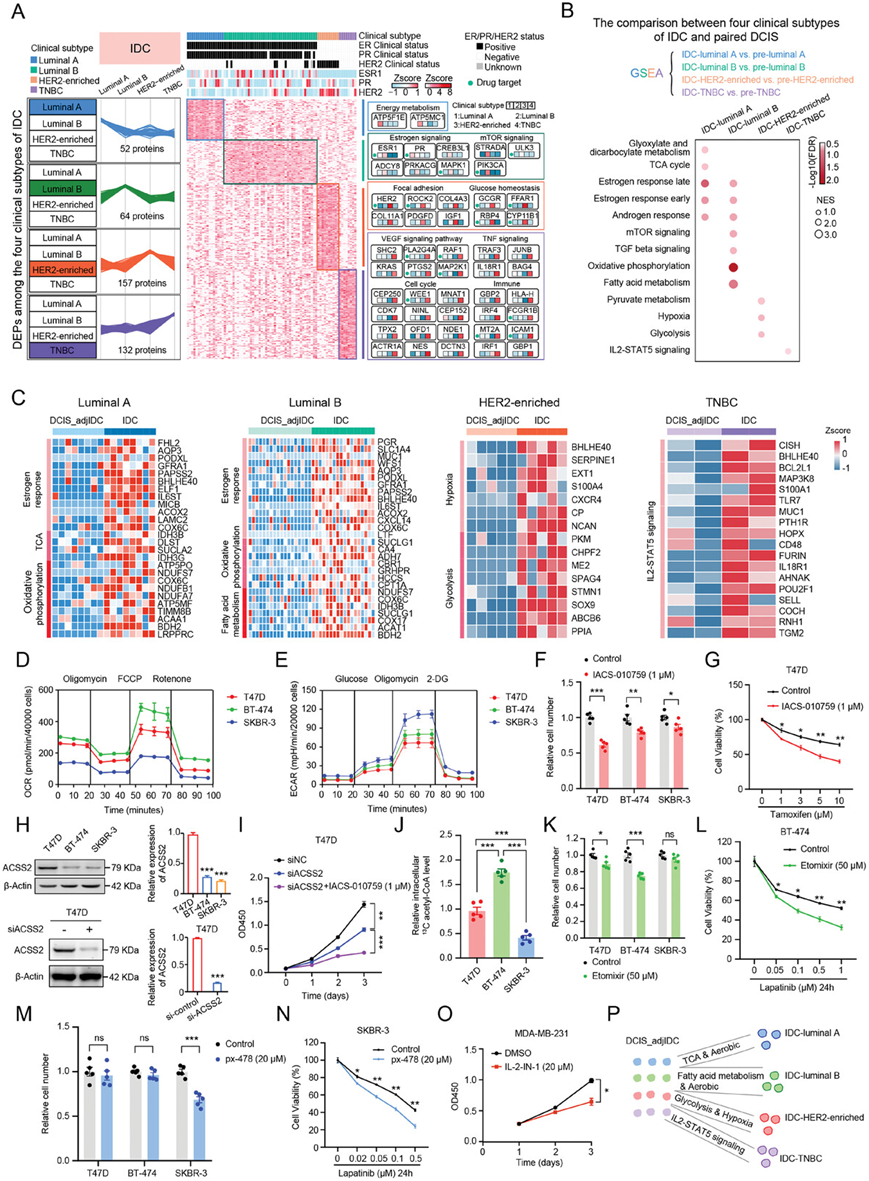
图7. DCIS 向 IDC 四种临床亚型转变的进展路线。
(A) 热图显示 IDC 四种临床亚型之间的蛋白质组差异。(B) 对四种临床亚型的 IDC 和配对的 DCIS 样本进行 GSEA。(C) 热图分别显示四种临床亚型的 IDC 与其配对的 DCIS_adjIDC 相比上调的通路和蛋白质。(D) T47D、BT-474 和 SKBR-3 细胞中的耗氧率 (OCR)。(E) T47D、BT-474 和 SKBR-3 细胞中的细胞外酸化率 (ECAR)。(F) OXPHOS 抑制剂 IACS-010759 (1µm) 显著抑制了 T47D 和 BT474 细胞的细胞增殖。(G) 他莫昔芬和 OXPHOS 抑制剂 IACS-010759(1 µ m)联合使用可抑制 T47D 细胞生长。(H) 免疫印迹分析。(I) 使用 siRNA 敲除 ACSS2 可抑制 T47D 细胞生长,而使用 IACS-010759 (1 µ m) 可对 siACSS2 细胞产生更显著的抑制效果。(J) 13C标记乙酰辅酶 A 相对水平。(K) Etomixir (50 µm) 显著抑制 BT-474 细胞增殖,但不抑制 T47D 和 SKBR-3 细胞增殖。(L) 拉帕替尼和Etomixir (50µm) 联合使用可抑制 BT-474 细胞生长。(M) HIF-1α 抑制剂 px-478 (20 µm) 抑制 SKBR-3 细胞增殖。(N) 拉帕替尼和 px-478 (20µm) 联合使用可抑制 SKBR-3 细胞生长。(O) IL-2 抑制剂 IL-2-IN-1 可显著抑制 MDA-MB-231 细胞的生长。(P) 该图显示了从 DCIS 到 IDC 的四种临床亚型的进展路径。
+ + + + + + + + + + +
结 论
本项研究对来自 168 例恶性和良性乳腺疾病患者的 224 个样本进行了全面的多组学分析,揭示了 BRDC进展的特点,例如TP53 突变相关ESR1 过表达参与了从导管增生到导管原位癌的转变。NR3C1过表达有助于 DCIS_Pure细胞避免免疫破坏。TIAM1-AR-AKR1C1轴促进 DCIS_adjIDC中的细胞侵袭和迁移。此外,AKR1C1 可作为潜在的治疗靶点,并证明了阿司匹林和地屈孕酮作为其抑制剂对肿瘤细胞的抑制作用。
+ + + + +



