

 English
English文献解读|Nat Commun(16.6):患重症疟疾贫血儿童的外周血转录组研究
✦ +
+
论文ID
原名:Entire expressed peripheral blood transcriptome in pediatric severe malarial anemia
译名:患重症疟疾贫血儿童的外周血转录组研究
期刊:Nature Communications
影响因子:16.6
发表时间:2024.06.12
DOI号:10.1038/s41467-024-48259-4
背 景
疟疾仍然是全球重大的公共卫生挑战,每年有 2.49 亿病例和 608000 人死亡。大多数病例(2.33 亿,93.6%)和死亡(580000,95.4%)发生在世卫组织非洲区域,由恶性疟原虫感染引起,主要发生在五岁以下儿童中。在高传播地区,严重疟疾的主要表现是存在或不存在呼吸窘迫的严重疟疾性贫血(SMA)[血红蛋白(Hb)<6.0 g/dL],脑型疟疾仅在罕见(非典型)病例中发生。
实验设计
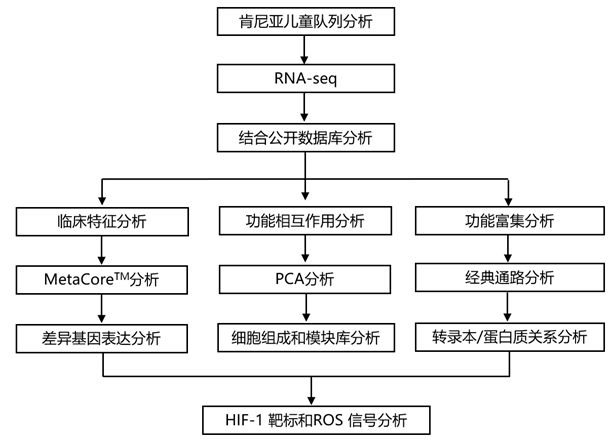
结 果
01

非 SMA 和 SMA 中的细胞通路比较揭示了不同的代谢和免疫反应
本项研究利用了 1644 名儿童(3-48 个月)队列的数据,这些儿童在 36 个月内进行了随访,在此期间研究团队记录了超过 19000 次血红蛋白测量值。由于儿童 SMA 是肯尼亚西部贫血相关死亡的主要原因之一,他们利用动态规划算法确定 36 个月随访期间的平均血红蛋白浓度,以确定最能反映儿童死亡率的类别。该方法在p < 0.001 时揭示了三个不同的血红蛋白类别:≤5.90 g/dL(n = 62,死亡率分数 = 0.53)、5.91-8.09 g/dL(n = 209,死亡率分数 = 0.15)和 >8.09 g/dL(n = 1373,死亡率分数 = 0.03)(图 S1)。从整个队列中选择了39名非SMA儿童和18名SMA儿童,不包括镰状细胞病(HbSS)儿童,以进行转录组分析(RNA-seq)。
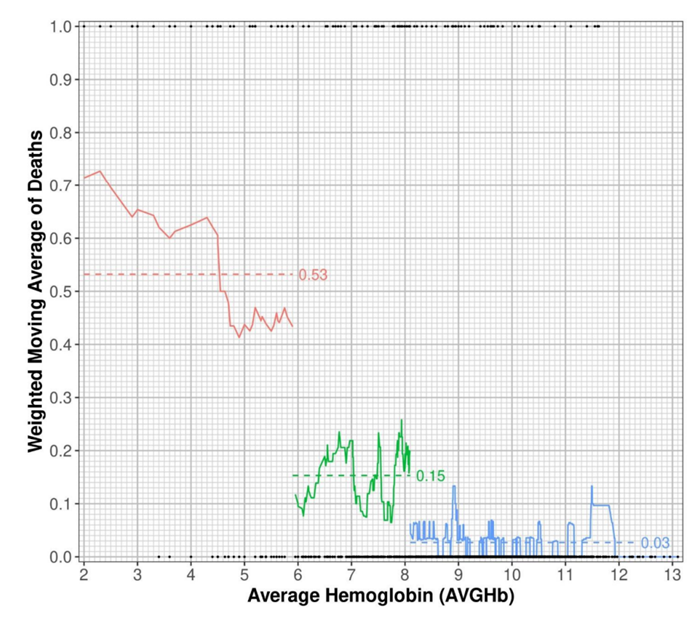
图S1. 按平均血红蛋白段计算的加权死亡率移动平均值。
为了识别独特和共有的基因,对 53286 个转录本进行了维恩图分析,发现在非 SMA 中独特表达的基因为 602 个,在 SMA 中为 493 个,还有 16036 个共同表达的基因(图 1a)。然后通过典型通路分析(MetaCore TM)探索非 SMA 和 SMA 中独特表达的基因,揭示了四个重要的子网络。非 SMA 儿童中排名最高的子网络是 [TFF3↔IL-6↔IL6RA↔ADAM17↔gp130],强调了t -辅助性17细胞系特化和分化、白细胞介素6介导的信号传导和T-辅助性细胞谱系承诺在驱动T-辅助性17型免疫反应中的关键作用的生物过程。第二个子网络 [SHOX2↔Neuregulin1↔FGFR2↔Endothelin-1↔ECE2] 表示调节细胞信号传导和增殖的过程,特别是通过受体酪氨酸激酶和酶联途径。总的来说,这些数据表明,非 SMA 的特征是免疫反应和细胞信号传导的变化,而 SMA 涉及更广泛的细胞活动,包括代谢调节和激素反应。然后对 53286 个转录本进行差异表达分析,确定了 1682 个差异表达基因(DEG):SMA 儿童中有 1403 个上调基因和 279 个下调基因(图 1b)。
主成分分析(PCA)显示,第一个主成分 (PCA1) 占总方差的 36.1%,区分了部分非 SMA 组和 SMA 组,而 PCA2(占总方差的 13.8%)对非 SMA 组具有更紧密的聚类(图 S3)。非 SMA 组在年龄、性别和镰状细胞特征方面表现出更紧密的聚类(同质性)。相比之下,SMA 组表现出更大的分散性,尤其是在男性和具有 HbAA 特征的人群中。因此,虽然有明显的基因表达特征可以区分这两组,但 SMA 儿童的异质性更强。
为了进一步探索 SMA 的发病机制,他们对显著的 DEG进行了非监督的层次聚类分析。这些分析确定了三个独特的共调控基因聚类,这些基因聚类将非 SMA 儿童和 SMA 儿童区分开来,它们的白细胞和淋巴细胞模式不同,但寄生虫血症水平、镰状细胞状态(HbAA 与 HbAS)或年龄分布不同(图 1c)。为了进一步了解主要共调控基因聚类的网络,生成了直接功能相互作用的典型通路图。SMA 中下调基因(聚类1)的网络为 IRF1↔SUZ12↔IL-1β↔NRF2↔LHX2,其中转录因子 IRF1 为中央发散枢纽(绿色方框,56 个直接相互作用),IL-1β 为中央汇聚枢纽(红色方框,44 个直接相互作用)(图 1d)。与聚类 1 的功能相互作用相关的过程是调节先天免疫反应和防御反应。聚类 2(上调基因)产生了一个 TAL1↔LYL1↔BRD4↔FOXO3A↔EKLF1 网络,其中转录因子 TAL1 为中心发散枢纽(绿色方框,343 个直接相互作用)。聚类 2(红色方框)中的中心汇聚枢纽是转录因子 E2F2(7 个功能相互作用),而次级发散枢纽是 GLUT1 和 HMBS(均具有 7 个直接相互作用)(图 1e)。聚类 2 的相互作用是代谢过程和红细胞分化。总的来说,SMA 与抑制先天免疫应激反应、增强代谢过程和促进红细胞分化有关。
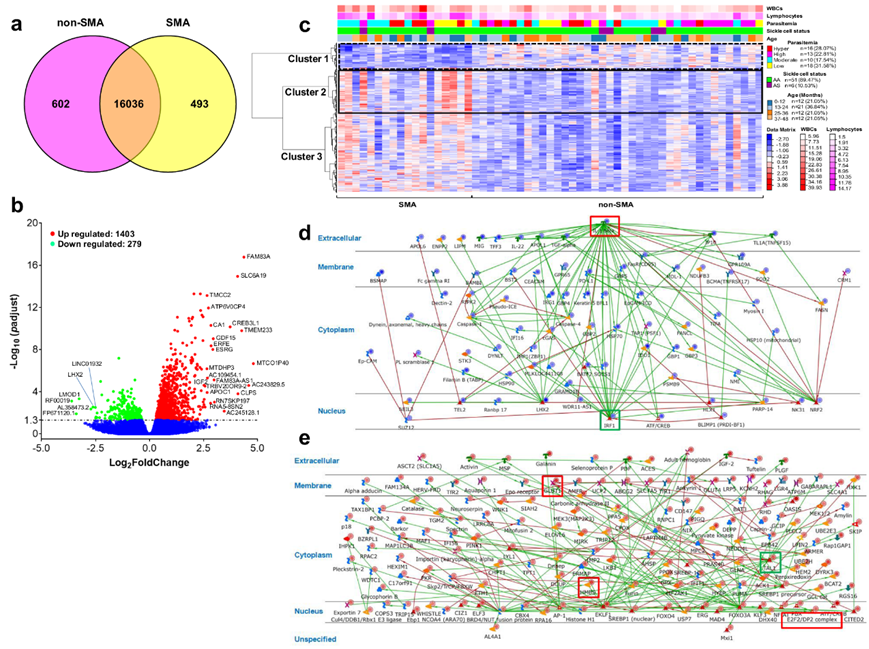
图1. 重症疟疾贫血儿童的差异表达基因分析。
(a) 维恩图描绘了临床组之间 53,286 个转录本的关系。 (b) 火山图显示,在肯尼亚患有非SMA (Hb≥6.0 g/dL, n = 39)和SMA的儿童中,1403个蛋白编码基因上调,279个蛋白编码基因下调。(c) 1682个差异表达基因的层次聚类热图。(d-e) 基于从热图中划分出聚类,突出显示了显著的基因网络和中央发散/汇聚中心。
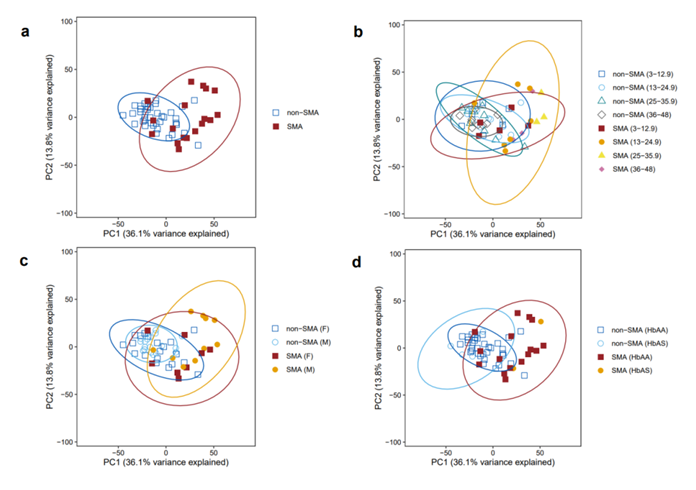
图S3. 非SMA和SMA的主成分分析(PCA)。
02
SMA 患者的白细胞免疫细胞谱发生改变
为了确定患有SMA的儿童的白细胞免疫谱是否不同,使用CIBERSORTx实现了一种生物信息学方法。尽管存在个体差异,但转录谱分析确定了五种免疫细胞类型,它们的表达差异为p < 0.050(图2a)。SMA 患儿的幼稚 B 细胞和 CD8 T 细胞表达增加(图2b)。相反,SMA 组的记忆 B 细胞、活化树突状细胞和中性粒细胞的表达比例较低(图2b)。根据表达数据中的免疫细胞类型谱,SMA 患儿的抗原反应似乎降低,免疫启动减少。
为了进一步确定与SMA相关的宿主免疫学特征,他们使用BloodGen3Module平台检测血液特异性基因表达特征。上调基因模式主要包含红系细胞(例如HBBP1、RHD、ST3GAL1、SLC2A1和GCLC)、蛋白质修饰(例如自噬和泛素-蛋白质组系统:FURIN、GABARAPL2、TM7SF2、RNF14、UBE2M、TAX1BP1)和中性粒细胞活化(例如CEACAM6、CEACAM8、CTSG、DEFA3、DEFA4)。相反,下调最多的基因特征是炎症(例如FCGR3B、TLR1、TLR5、FPR2、HIF1A、SOD2、HSD17B11、MAPK14、SOCS3、LIMK2、ACSL1、NDUFB3)、干扰素和细胞因子/趋化因子(CSF2RB、FPR2、IFIT2、IFIT5、MX1、NOD2、GBP1、GBP4、GBP5、CASP1、CASP4、CASP5、MAPK14、TAP1、PSMB9、SP100)和中性粒细胞(例如HSPA1A、ADAM8、FCGR2A、FCGR3B、FPR1、CYB5R4、NCF4、HLA-E、LCP1、BCL3、IRF1、NFKBIA、BCL3和MCL1)(图2c-e)。
总之,SMA的特点是红系细胞上调,中性粒细胞活化增强,炎症反应受损。
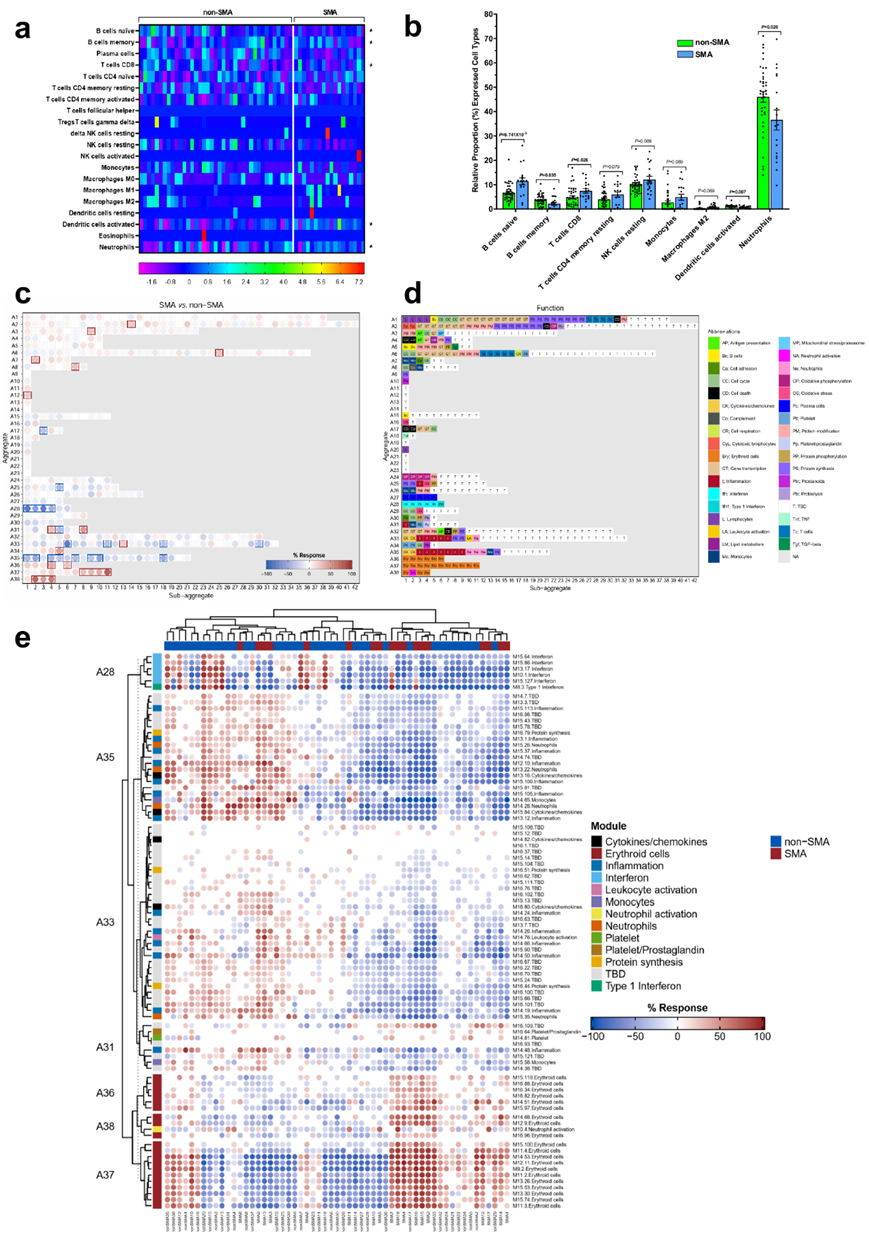
图2. 全血的细胞组成和模块库分析。
(a) 热图表示非 SMA(Hb≥6.0 g/dL)和 SMA(Hb<6.0 g/dL)组中个体患者水平上 22 种类型/亚型白细胞群的细胞类型表达 。(b) 非SMA和 SMA组之间不同免疫细胞类型的相对表达比例(%)。(c) 模块指纹网格图分析结果。每个模块都位于一个网格上,行对应于“模块聚合体”,反映不同免疫状态的参考数据集中的相似丰度模式。(d) 网格内每个模块相关的生物/免疫功能。(e) 指纹热图表示跨个体研究参与者的注释模块模式。
03
功能富集分析
为了确定与 SMA 发展相关的特征,对三个领域(即生物过程、细胞成分和分子功能)进行了GO富集分析(图3a)。在这三个领域中,生物过程在 SMA 儿童中表现出最显著的富集。例如,蛋白酶体介导的泛素依赖性蛋白质分解代谢过程、原卟啉原 IX 代谢过程、和自噬调节是排名靠前的生物过程。泛素连接酶复合物、吞噬泡组装位点和血红蛋白复合物是 SMA 中富集最多的细胞成分。染色质 DNA 结合、蛋白质-大分子接头活性和转录辅阻遏物活性显示出分子功能的最大富集(图3a)。
使用 Reactome 富集进行的额外分析发现,磷脂代谢、BH3 纯蛋白的激活和血红素生物合成是 SMA 患儿中排名靠前的通路(图3b-c)。总之,富集分析表明,SMA 的特征是多方面的紊乱,涉及蛋白质降解(泛素化)、血红素代谢、细胞清除机制(自噬)和胞吐作用,与细胞稳态和调节通路的破坏一致。KEGG 通路的 DEG 功能分类揭示了三条重要途径:胞吐作用、线粒体自噬和细胞自噬(图S4)。总之,富集分析表明,SMA 的特征是多方面的紊乱,涉及蛋白质降解(泛素化)、血红素代谢、细胞清除机制(自噬)和胞吐作用,与细胞稳态和调节通路的破坏一致。
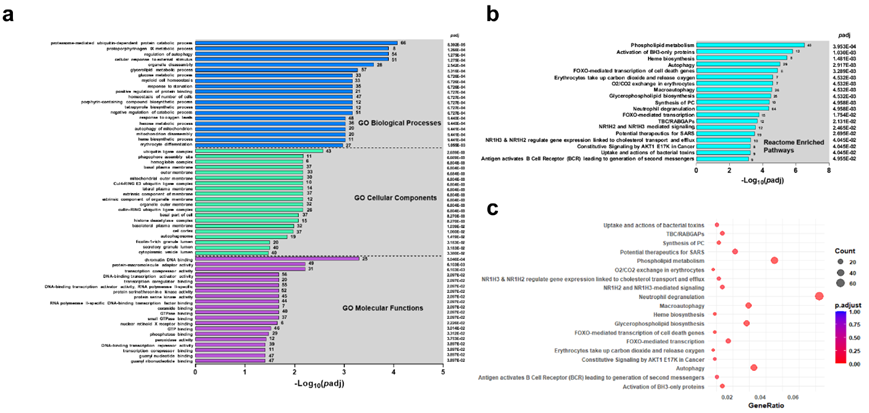
图3. 重症疟疾贫血差异表达基因的功能富集分析。
(a)GO富集分析。(b) 对SMA患儿中存在显著差异的 19 个富集项进行反应组富集分析。(c) Reactome富集直方图。
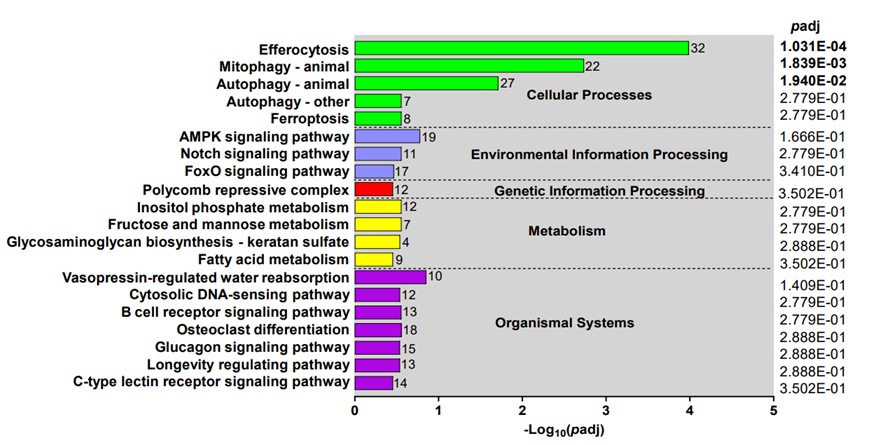
图S4. DEG的KEGG通路功能分类。
04
经典通路聚焦于 SMA 中的细胞应激反应、免疫调节和代谢变化
通过探索前 10 条显著的典型通路图(MetaCore TM)对 SMA 发病机制进行了进一步表征(图 4a)。功能类别中排名靠前的通路是:(i)细胞凋亡和存活——线粒体蛋白调节细胞凋亡;(ii)免疫反应——通过 MAPK 进行 IFN-α/β 信号传导;(iii)氧化应激——ROS 信号传导;(iv)代谢调节——葡萄糖和脂质代谢中的糖皮质激素受体信号传导; (v) 信号转导 - mTORC2 下游信号传导;以及 (vi) 转录 - HIF-1 靶标。这些通路共同揭示了 SMA 中细胞应激反应、免疫调节、代谢变化和转录调控之间的复杂相互作用。
为了验证 RNA-Seq 结果,他们利用了来自乌干达 SMA 儿童队列和社区儿童(对照2的转录组数据集。使用 323 个基因对通路图进行富集分析,发现参与转录的 HIF-1 靶标为最重要的新兴通路(图 S5),这是数据集中排名第二的通路图。因此,两个数据集都映射到 HIF-1 靶标通路上,以直接比较 DEG(图4b)。在乌干达数据集的前 10 条通路中(图 S5),肯尼亚数据集中唯一的其他常见通路是参与氧化应激的 ROS 信号传导。
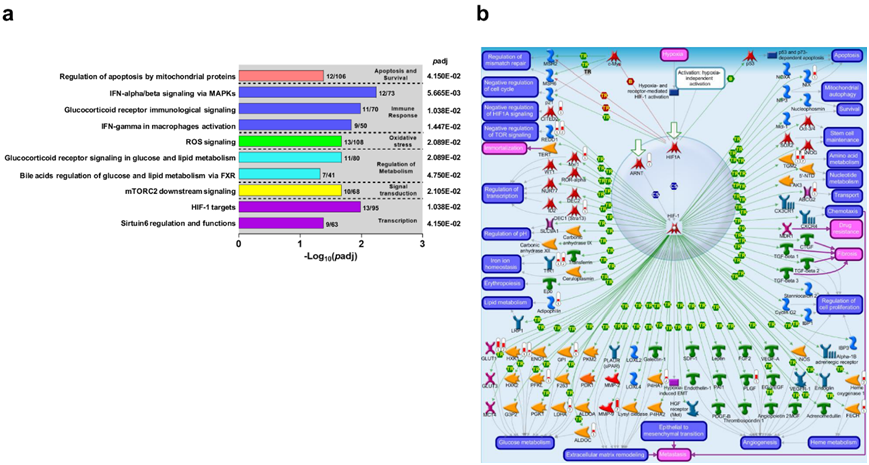
图4. 严重疟疾贫血中排名靠前的 Metacore TM典型通路图。
(a) 从 RNA-Seq 分析中出现的排名前 10 的经典通路。(b) 经典通路图。
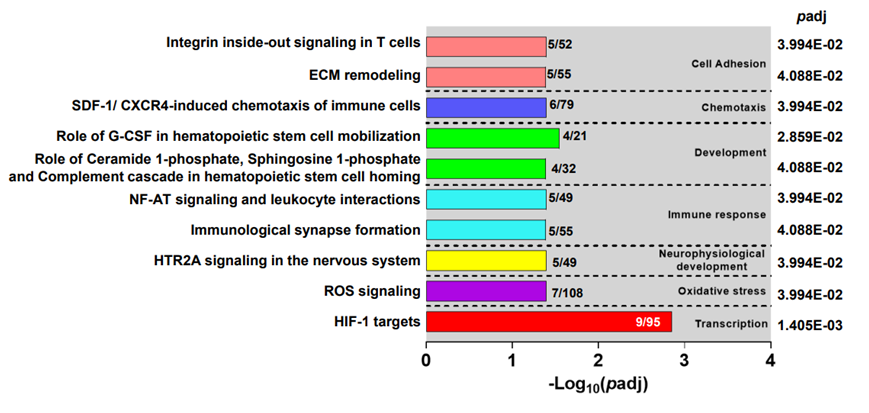
图S5.乌干达数据集的排名靠前的MetacoreTM典型通路图数据。
05
使用外部数据集和靶向 RNA-Seq 验证全血转录组数据
为了进一步验证全血转录组分析中确定的 DEG,他们在单独的肯尼亚儿童队列[非 SMA(n = 23)和 SMA(n = 20)] 中利用了 491 个免疫反应基因的 Qiagen 靶向 RNA-seq。热图聚类分析显示两个平台的方向性一致(图 5a),两个数据集显示出稳健的相关性(图 5b)。外部数据集和靶向 RNA-seq验证与肯尼亚儿童的整个转录组学具有高度一致性,表明不同 SMA 队列之间的 DEG 一致。
为了确定 RNA-Seq 中的 DEG 是否与蛋白质水平的变化一致,他们将全血转录组数据与蛋白质丰度进行了比较。MetaCore TM用于将 35 名儿童的基因映射到各自的蛋白质产物,这 35 名儿童同时具有两种测量值。对 405 个基因/蛋白质对的分析显示热图和散点图一致,且呈中等正相关(图 6a-b)。然后在 MetaCoreTM中生成通路图,从而可以全面了解所有转录本/蛋白质关系。最一致的通路是 HIF-1 靶标,在转录本和蛋白质中均表现出高度显著性,表明转录组和蛋白质丰度变化之间存在一致的生物学重叠,尤其是对于 HIF-1 相关过程(图 S7)。总之,这些数据表明转录本和蛋白质测量的幅度和方向性之间存在适度的关系,对于 HIF-1 靶标,两个分子层上的生物学变化模式一致。
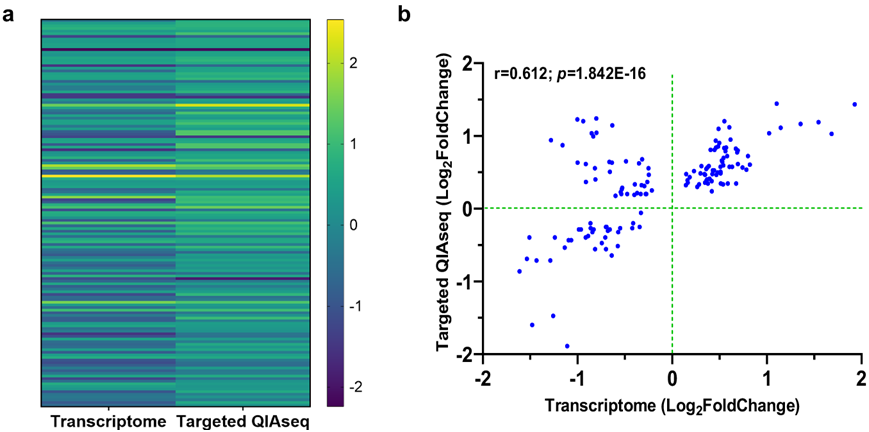
图5. 使用靶向 RNA 测序面板验证全血转录组数据。
(a) 热图说明了两个数据集之间显著 DEG的比较 。(b) 相关散点图展示了靶向 QIAseq 分析与转录组数据中显著表达的基因之间的关系。
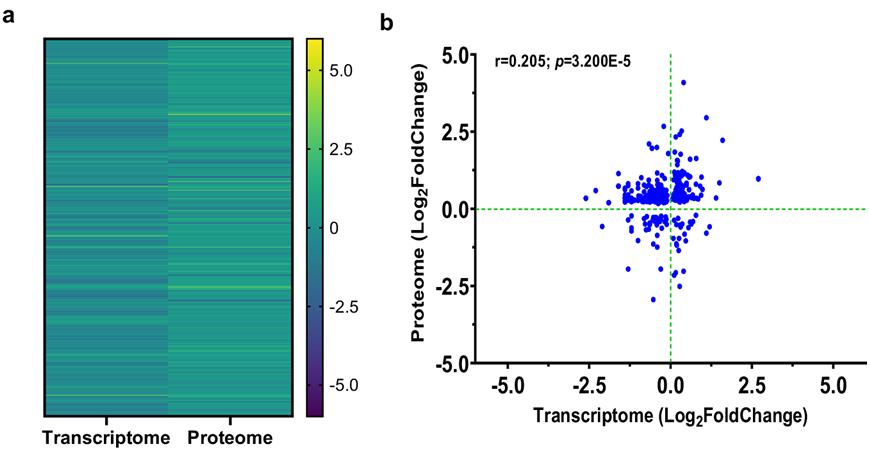
图6. 转录组学和蛋白质组丰度的比较分析。
(a) 热图显示两个数据集之间显著基因/蛋白质对的比较 。(b) 相关散点图展示了显著表达的蛋白质靶标和基因之间的关系。
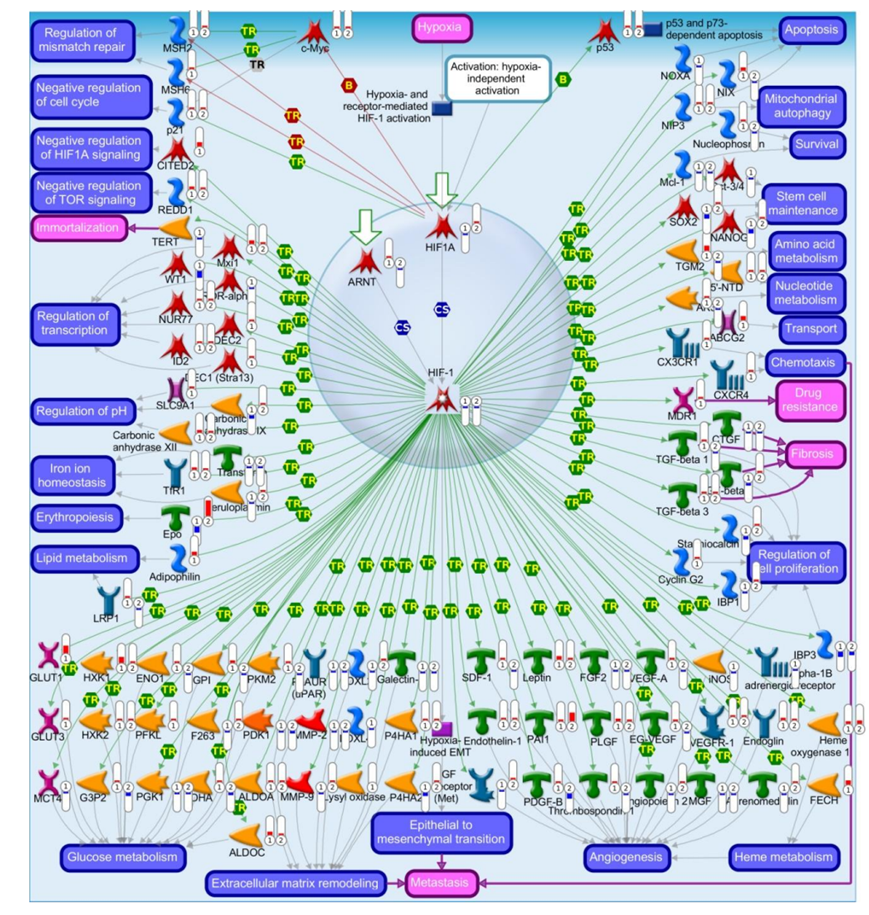
图S7. 使用MetacoreTM经典通路图对转录本和蛋白丰度进行比较分析。
+ + + + + + + + + + +
结 论
本项研究比较了肯尼亚儿童中非 SMA和 SMA之间的转录组数据。差异表达分析显示 SMA 中有 1403 个上调转录本和 279 个下调转录本,表明宿主炎症小体活化、细胞死亡以及先天免疫和细胞应激反应受损。免疫细胞分析显示记忆反应、抗原呈递和即时病原体清除减少,表明 SMA 中的免疫反应不成熟/调节不当。血液特异性基因特征的模块库分析确定了 SMA 中的红细胞基因上调、中性粒细胞活化增强和炎症反应受损。富集分析集中于细胞稳态破坏和泛素蛋白酶体系统、自噬和血红素代谢的调节通路。途径分析强调了在缺氧条件下激活 [缺氧诱导因子 (HIF)-1 靶标和活性氧 (ROS) 信号] 是 SMA 的关键通路,这些信号通路在蛋白质丰度测量和具有可用转录组数据的乌干达 SMA 队列中也排名靠前。这些结果确定了 SMA 发病机制中的关键分子主题,为新的疟疾疗法提供了潜在靶点。
+ + + + +



