

 English
English文献解读|Nature(48.5):母体肥胖导致库普弗细胞编程引发脂肪肝
✦ +
+
论文ID
原名:Kupffer cell programming by maternal obesity triggers fatty liver disease
译名:母体肥胖导致库普弗细胞编程引发脂肪肝
期刊:Nature
影响因子:48.5
发表时间:2025.07.18
DOI号:10.1038/s41586-025-09190-w.
背 景
早期环境因素对成年期患非传染性疾病的风险有很大影响,这一概念称为健康和疾病的发育起源 (DOHaD)。母亲营养不良、肥胖、感染、环境污染物和社会心理压力与后代的代谢紊乱、神经系统疾病、癌症和免疫反应失调有关。在细胞水平上,母体免疫激活可以改变胎儿造血干细胞和祖细胞 (HSPC),导致新生儿败血症。母亲高脂饮食 (HFD) 或病毒感染也会对脑内驻留巨噬细胞小胶质细胞进行编程,导致神经元疾病。然而,脑外巨噬细胞是否可以充当代际信使并与疾病的发生有因果关系,作为 DOHaD 中的细胞成分,仍然不清楚。库普弗细胞 (KC) 是组织驻留巨噬细胞,在胚胎发生早期定植于肝脏。在肝脏定植后,KC 迅速获得组织特异性转录特征,与发育中的肝脏一起成熟并适应其功能。在整个发育和成年期,KC 执行对肝脏和生物体稳态至关重要的核心功能,包括支持胎儿红细胞生成、出生后红细胞循环和肝脏代谢。然而,发育过程中巨噬细胞核心功能的扰动是否会导致或引起出生后疾病尚不清楚。
实验设计
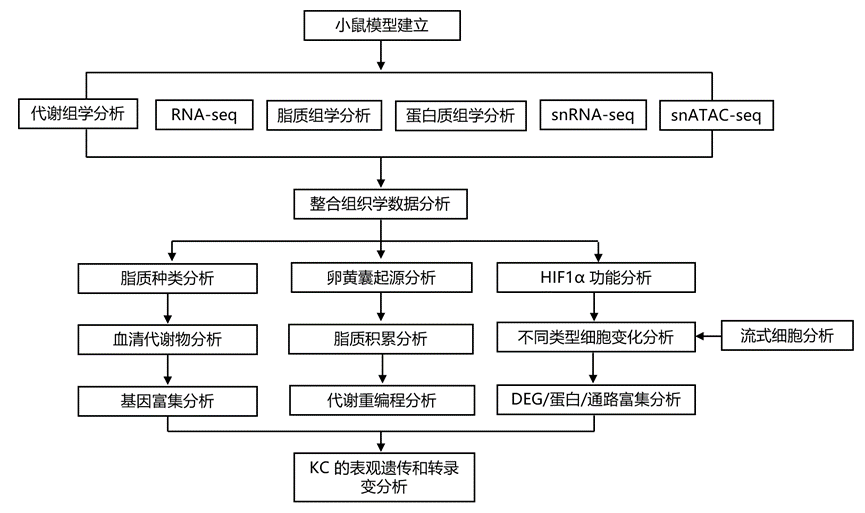
结 果
01

孕妇肥胖导致脂肪肝
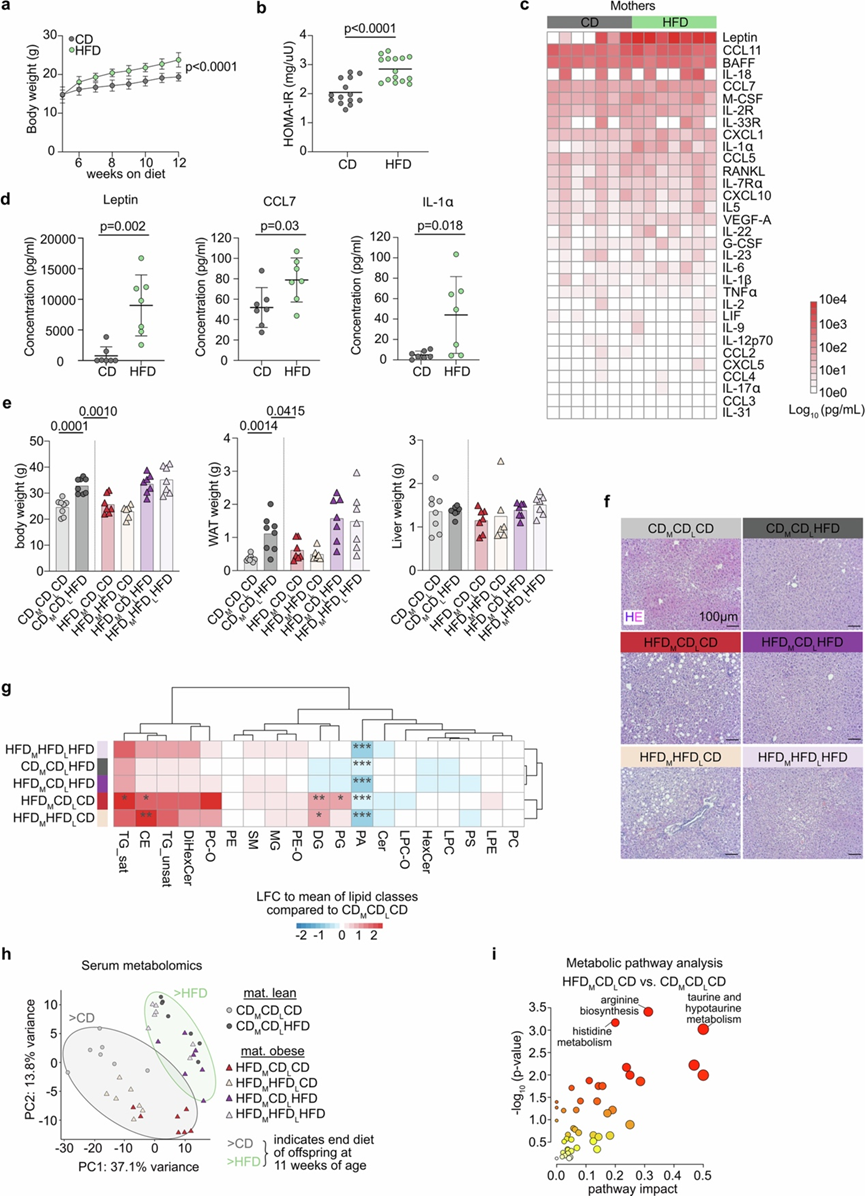
在人类和动物模型中,母亲肥胖与后代肥胖和肝病有关,但其机制仍不清楚。他们假设 KC可能由于其早期肝脏定植和代谢作用而导致脂肪肝 (FLD)。为了验证这一假设,用高脂饮食(HFD)喂养 C57BL/6Jrcc 雌性小鼠 8 周,导致体重增加和胰岛素抵抗的稳态模型 (HOMA-IR) (图S1a-b)。通过血清细胞因子和趋化因子分析评估,这些饮食条件不会导致母体炎症,只有白细胞介素 1α (IL-1α)、CC 基序趋化因子配体 7 (CCL7)和瘦素上调(图S1c-d)。然后将雌性与对照饮食 (CD) 喂养的雄性进行交配。为了分析妊娠期与哺乳期的影响,将新生小鼠交叉哺乳和/或断奶后喂食CD或HFD(母体肥胖组)(图1a)。作为母体瘦对照组,他们分析了CD雌性所生的后代,这些后代终生以CD喂养或以HFD断奶(图1a)。在11周时,出生后以CD喂养(HFDMCDLCD;M表示妊娠期母体饮食,L表示哺乳期寄养母体饮食)或断奶(HFDM HFDLCD)的母体肥胖组与以CD喂养的母体瘦弱组(CDMCDLCD)相比,体重或白色脂肪组织(WAT)重量没有差异(图S1e)。只有在出生后采用 HFD 的组(CDMCDLHFD、HFDMCDLHFD 和 HFDM HFDLHFD)才会出现肥胖,这表明断奶后的饮食(而不仅仅是母亲的肥胖)决定了整体体重和 WAT 重量(图S1e)。
相反,油红O(ORO)和苏木精和伊红(H&E)染色显示,所有母体肥胖组的肝脏脂质含量增加,虽然在所有条件下肝脏重量相似(图1b-c,图S1e-f)。主成分分析(PCA)和脂质组学分析的层次聚类均表明,以CD作为最终饮食的母体肥胖组(HFD> CD)与所有以HFD为最终饮食的动物(CD>HFD和HFD>HFD)不同,所有这些都与CD M CD L CD(CD>CD)不同(图1d)。事实上,HFDMCDLHFD和 HFDM HFDLCD组显示出脂质种类显著增加,例如饱和三酰甘油、二酰甘油和胆固醇酯,而与CDMCDLCD肝脏相比,CDMCDLHFD以及母亲肥胖 HFDM CDLHFD 和 HFDMHFDLHFD 组仅显示出脂质积累适度增加。
接下来,他们对 900 多种代谢物和脂质进行了血清代谢组学分析,以检测肥胖母亲所生后代的 FLD 表型是否也与系统性改变有关,这些改变甚至可以作为特定的生物标志物。组间差异主要反映最终的饮食状况(CD 与 HFD)(图S1h)。比较 CDMCDLCD 与 HFDM CDLCD 后代,其中身体成分相似,但 FLD 表型在 HFDMCDLCD 肝脏中最为突出,揭示了 129 种显著改变的代谢物,它们在代谢通路“牛磺酸和亚牛磺酸代谢”,“精氨酸生物合成”和“组氨酸代谢”中富集(图S1i)。具体而言,谷氨酰胺:谷氨酸比率(参与精氨酸生物合成的两种氨基酸)在HFDMCDLCD小鼠中显著升高。虽然FLD中该比率普遍降低,但母体肥胖模型报告称,该比率升高与肝脏代谢状态相关,提示该比率可能是母体肥胖导致的FLD的生物标志物。总而言之,与出生后饮食诱导的FLD相比,母体肥胖会在后代中诱导出不同的FLD表型。
接下来,他们重点研究了肝脏髓系细胞,以了解母亲肥胖引起的 FLD 表型,这些细胞在 FLD 的起始和传播中起着关键作用。定量分析证实了 HFD M CD L CD 后代中中性粒细胞、单核细胞和经典树突状细胞(cDC1 和 cDC2)增加。母亲肥胖组中的 KC 数量增加,而肝囊巨噬细胞不受影响。母亲瘦肉对照组 (CDMCDLHFD) 断奶后 HFD 未诱导髓系细胞内流。对分选的KC进行的转录组分析(RNA-seq)显示,其聚类模式取决于宫内暴露于瘦型(CD>)或肥胖型(HFD>)母体环境(图1e)。共表达网络分析(hCo-Cena)确定了五个模块(图1f),蓝色模块代表因母亲肥胖而上调的基因。使用GO、KEGG和 HALLMARK 数据库进行的通路富集分析显示,与“参与免疫反应的细胞活化”相关的基因上调。其中包括Trem2,该基因在 FLD期间在 KC 中上调,并且在同一模块中伴随其下游信号级联分子Tyrobp和Syk(图1g)。此外,Myd88、Gpnmb和单核细胞特异性基因Ccr2和Cx3cr1在蓝色模块中,在 HFDMCDLCD KC中表达最高(图1g)。他们发现“糖酵解/糖异生”在蓝色模块中富集(图1f)。相比之下,青绿色和浅绿色模块表明“氧化磷酸化”、“细胞呼吸”和“外来化合物代谢”等代谢过程在母体肥胖时下调(图1f)。因此,母体肥胖会引起 KC的炎症反应,并导致其在转录水平上的代谢状态改变。为了验证 KC 在蛋白质水平上的转录变化,他们对来自 CDMCDLCD 和 HFDMCDLCD 后代的 KC进行了基于光谱流式细胞的代谢分析。虽然参与葡萄糖和脂质代谢通路的大多数营养转运蛋白和代谢酶保持不变,但与CDMCDLCD 相比,HFDMCDLCD KC中的琥珀酸脱氢酶 (SDHA)(三羧酸循环的关键组成部分)和葡萄糖转运蛋白 1 (GLUT1) 有所降低。这些发现表明,母体肥胖会损害子代 KC的代谢能力。
图S1. 母鼠肥胖模型和后代肝脏代谢状态的特征。
(a) 体重分析。(b) 在相应饮食 8 周后对 CD 或 HFD 母亲进行的胰岛素抵抗稳态模型评估 (HOMA-IR)。(c) 热图显示 CD 和 HFD 母亲血清中检测到的细胞因子和趋化因子的对数转换值。(d) (c)中结果的绘图,使用双尾非配对学生 t 检验显示其显著性。(e) 11 周龄后代体重、白色脂肪组织 (WAT) 和肝脏重量。(f) 后代肝脏切片的苏木精-伊红 (HE) 染色。(g) 肝脏脂质组学热图显示了不同条件下所选脂质组的CDM CDLCD的变化。(h) 通过 PCA 可视化所有实验组血清代谢组学数据。(i) 代谢通路富集分析。
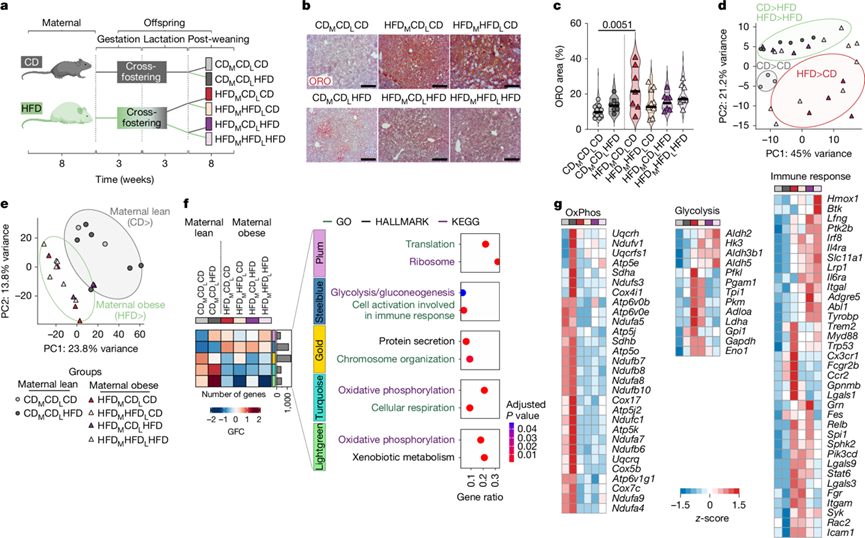
图1. 母鼠肥胖导致后代发生 FLD。
(a) 母体肥胖小鼠模型的产生。(b) 子代肝脏的 ORO 染色。(c) 通过 QuPath 对(b)的 ORO 染色进行定量。(d) 通过 PCA 可视化所有实验组的肝脏脂质组数据。(e) 通过 PCA 可视化排序的 KC 的RNA-seq 数据。(f) 共表达网络分析。 (g) (f)中 DEG 与相应注释基因集交集的选定基因的热图。
02
KC保留了卵黄囊起源
饮食诱导的脂肪肝(FLD)模型显示单核细胞浸润增加并替代卵黄囊来源的库普弗细胞(KCs)。RNA-seq数据提示母体肥胖子代KC中存在单核细胞特征。为探究KC的个体发育,他们构建了Tnfrsf11aCre;Rosa26 LSL-YFP;Ms4a3 FlpO;Rosa26 FSF-tdTomato双谱系示踪小鼠,通过重组酶系统同步追踪卵黄囊来源的前巨噬细胞与骨髓来源单核细胞的发育谱系(图2a-b)。既往证实Tnfrsf11aCre小鼠能高效标记前巨噬细胞及其后代,但由于Tnfrsf11a是巨噬细胞核心基因,单核细胞分化为巨噬细胞时会激活其表达,导致无法区分胎儿源与骨髓源巨噬细胞。因此,他们同时采用新开发的Ms4a3FlpO小鼠模型,该位点特异性标记单核细胞及所有单核细胞来源的巨噬细胞。验证显示Ms4a3FlpO的示踪效率与Ms4a3Cre模型相当,超过95%的Ly6C+血液单核细胞由tdTomato(tdT)标记。其中约13%单核细胞已表达YFP,提示这些细胞开始启动巨噬细胞核心程序,但未发现仅YFP+的单核细胞,证实该双示踪系统能明确区分卵黄囊来源与骨髓来源的造血细胞群。小胶质细胞作为卵黄囊来源细胞也显示高YFP标记率。在11-12周龄CDMCDLCD小鼠肝脏中,超过96%的KC为YFP+,仅极少数显示tdT标记(图2c),符合卵黄囊起源特征。虽然存在FLD表型和单核细胞基因上调,HFDMCDLCD组KC仍保持高YFP标记率(约96%)。这表明即使经历母体肥胖和成年期FLD环境,KC仍维持卵黄囊起源,可作为代际传递信使将宫内环境信息传递至成年阶段。
他们假设,母体肥胖对 KC的发育编程会削弱其稳态功能,并通过旁分泌信号传导促进 FLD。为了验证这一假设,他们将肝细胞与来自 CDMCDLCD 或 HFDMCDLCD 肝脏的 KC共培养,不进行直接细胞接触,确保仅发生旁分泌信号传导(图2d)。使用 LD540 标记监测肝细胞中的实时脂质积累,以通过活细胞成像可视化中性脂质。与和CDMCDLCD KC 共培养的肝细胞相比,与 HFDMCDLCD KC共培养的肝细胞表现出明显更高的脂质积累(图2e)。因此,在胚胎发育阶段受到母体肥胖饮食因素影响的KC,可通过旁分泌信号发挥其促进FLD的功能。
为进一步在体内验证KC依赖性肝细胞脂质积聚,他们采用了KC-白喉毒素受体(DTR)小鼠模型:该模型在KC特异性Clec4f启动子驱动下表达人源DTR,单次注射白喉毒素即可在24小时内实现KC的靶向清除。他们将HFDMCDLCD子代与Clec4fDTR/+或Clec4f+/+基因型交配。于出生后第0天(P0)给予白喉毒素以清除Clec4fDTR/+小鼠体内的KC,并于P1移植来自正常小鼠的tdT⁺骨髓单核细胞及HSPC,以重建KC生态位(图2f)。虽然成年Clec4fDTR/+小鼠肝脏中仍有大量来源于内源性tdT⁻祖细胞的KC样细胞,他们仍检测到tdT⁺ KC样细胞的存在,该处理显著逆转了Clec4fDTR/+ HFDMCDLCD小鼠的脂肪肝表型,Oil Red O及H&E染色显示肝细胞脂滴积聚减少(图2g-h)。相反,接受相同白喉毒素注射与细胞转移的Clec4f+/+同窝对照仍保留脂质积聚,与C57BL/6Jrcc小鼠结果一致(图1b-c)。此外,仅依靠内源性单核细胞、HSPC及残余KC重建空缺的KC生态位(即Clec4fDTR/+小鼠仅注射白喉毒素,不接受正常来源的细胞移植)并不能挽救脂肪肝表型(图2g-h),提示HSPC本身可能已发生发育性编程,与既往报道一致。综上,本研究证明了肥胖母鼠子代中,KC来源的旁分泌因子驱动肝细胞脂质积聚。
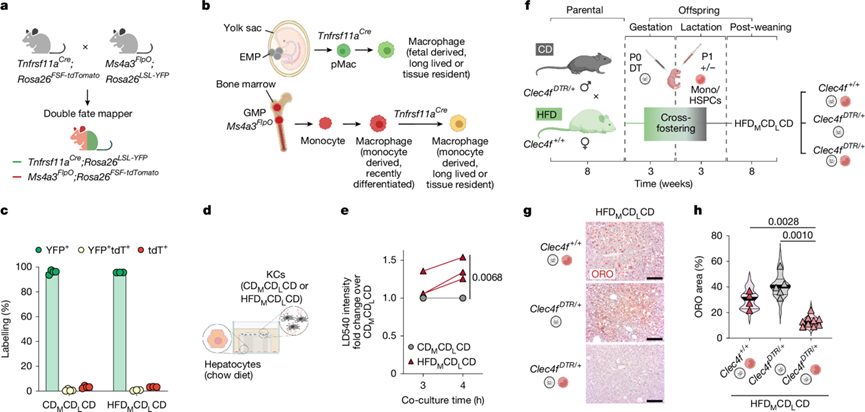
图2. 母亲肥胖后,KC仍保留卵黄囊起源,并通过旁分泌信号引起脂滴积聚。
(a-b) 实验方案。(c) 从小鼠中分离的KC中双谱系模型的标记效率 。(d) 从添加 KC 的饲料喂养小鼠中分离的肝细胞培养物示意图。(e) 通过活体成像,在与 CDM CDL CD 或 HFDMCDL CD 小鼠的 KC共培养 4 小时的体外培养肝细胞中以标准化 LD540 强度显示脂质积累。(f) KC耗竭和细胞转移母体肥胖模型的产生。(g) 子代小鼠的肝脏 ORO 染色。(h) ORO 染色进行定量分析。
03
HIF1α驱动KC编程
由于HIF1α调控巨噬细胞从氧化磷酸化到糖酵解的代谢转换,他们检测了通过母体肥胖阻止HIF1α依赖的KC发育编程是否能挽救FLD表型。他们使用了LysM Cre;Hif1a f/f小鼠,其中Hif1a在巨噬细胞和其他髓系细胞中敲除。由于哺乳期母体饮食的影响微乎其微(图1),他们重点关注了母体肥胖组出生后的饮食转换,并以断奶后HFD喂养的动物作为对照组(图3a)。当最终饮食为 HFD(CDMHFD 和 HFDMHFD)时,Hif1a f/f[野生型(WT)]和LysMCre;Hif1a f/f (KO)同窝仔鼠的体重和 WAT 重量均增加,这表明髓系特异性 HIF1α 缺失并不能预防饮食引起的肥胖。
接下来,他们关注肝脏表型。与 C57BL/6Jrcc 小鼠一样,肝脏总重量保持不变,肥胖母鼠 (HFDMCD WT)所生的Hif1a f/f后代出现了 FLD,这通过 ORO 染色(图3b-c)和脂质组学观察到(图3d)。与 C57BL/6Jrcc 小鼠的脂质组学结果相似,HFDMCD WT 后代的饱和三酰甘油和胆固醇酯增加。然而,HFD M CD KO 后代没有出现 FLD(图3b-d),这表明在胚胎发生过程中 KCs 中 HIF1α 的缺失足以阻止其代谢编程,最终导致脂质在肝细胞中积累。当KO后代出生后继续食用HFD(HFDMHFD KO)时,这种效应减弱,因为它们表现出与HFDMHFD WT类似的FLD发育,脂质积累较高(图3b-c),三酰甘油、二酰甘油、单酰甘油和胆固醇酯显著增加。
为了研究 KC中 HIF1α 依赖的发育程序如何导致 FLD,他们分析了来自母亲瘦和母亲肥胖条件的 WT 和 KO KC的转录组。母体肥胖而非终末饮食是KC转录程序的主要驱动因素(图3e),与C57BL/6Jrcc模型结果一致(图1e)。仅来自HFDMCD KO小鼠的KC与母体瘦条件聚类在一起(图3e),表明母体肥胖引起的发育性编程事件依赖于HIF1α。特别关注 HIF1α 依赖的基因调控,比较 HFDMCD WT 与 HFDMCD KO KCs,产生了 409 个 差异表达基因(DEG)。对HFDMCD KO KC中下调的DEG进行GO分析,发现了诸如“脂肪酸代谢过程”、“细胞对外来化合物代谢的反应”和“血液凝固、纤维蛋白凝块形成”等通路。由于体外培养数据表明KC通过旁分泌因子引起脂质积累(图2d-e),他们寻找与肝细胞受体相对应的分泌配体。在25种差异表达配体中,与HFDMCD KO KC相比,大多数在HFD M CD WT中上调(图3f)。其中,他们检测到了载脂蛋白(Apob和Apoa1)和许多凝血因子基因,例如Fga,Fgb,Fgg,F2,F8和F9,它们分别编码纤维蛋白原,凝血酶和凝血因子VIII和IX。
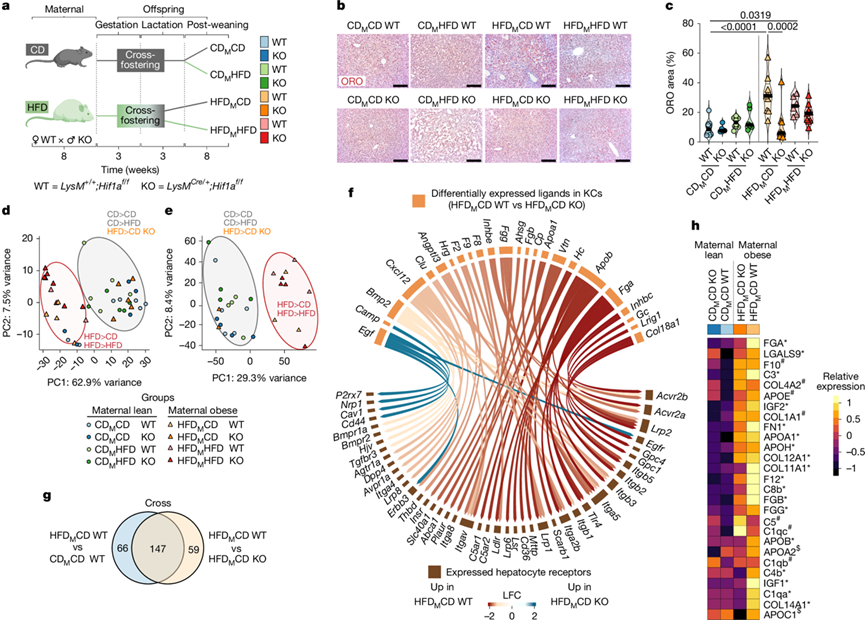
图3. 出生后 FLD 由 HIF1α 依赖的 KC 发育编程驱动。
(a) 通过髓系Hif1a敲除建立母体肥胖模型。(b) 代表性肝脏ORO 染色。(c) 通过 QuPath 对(b)的 ORO 染色进行定量。(d) 通过 PCA 可视化肝脏脂质组学数据。(e) 通过 PCA 可视化的RNA-seq数据。(f) 弦图显示肝细胞表达的KC 配体及其各自的受体。(g) 差异表达蛋白分析。(h) 热图显示(g)中选定的蛋白质。
04
KC的表观遗传和转录变化
为确定母体肥胖诱导的变化是否持续至成年,并避免分选KC可能带来的污染,他们对12周龄的C57BL/6Jrcc HFDMCDLCD和CDMCDLCD子代肝脏进行单细胞转录组分析(snRNA-seq)和单细胞转座酶可及染色质测序(snATAC-seq),该方法可在单细胞水平同时分析表观遗传和转录组特征。整合snRNA-seq与snATAC-seq数据后,获得了包括KC和肝细胞在内的多种肝实质细胞类型(图S9a-b)。对KC进一步亚聚类得到5种不同的KC状态(图4a-b),其中聚类0、1和4高表达Adgre1、Timd4和Clec4f。在HFDMCDLCD条件下,相对占比升高的聚类2和聚类3分别高表达MHC II类相关基因(H2-Aa、H2-Ab1和Cd74)(聚类2),以及免疫调控和代谢应答基因(Pparg、Lilr4b和Lgals3)(聚类3)(图4b)。HFDMCDLCD肝实质中MHC II类表达升高得到验证,而CDMCDLCD小鼠仅在肝门静脉及中央静脉周围呈现限制性MHC II类表达。
比较 HFDMCDLCD 和 CDMCDLCD KC之间的 snATAC-seq 峰相关基因,发现 28 个下调基因位点和 60 个上调基因位点(图S9d)。虽然数量相对较少,但下调基因的 GO分析表明脂质代谢过程的富集(图S9e)。值得注意的是,ATAC-seq 峰在Pck1基因座内存在差异,Pck1 基因座编码磷酸烯醇式丙酮酸羧激酶 1,这是一种通常对糖酵解具有拮抗作用的酶。绘制基因表达图验证了所有 HFDMCDLCD KC 状态下Pck1表达的丧失(图S9f),与之前描述的代谢转换一致(图1f-g)。此外,他们观察到Gpnmb基因座内的表观遗传变化,其中 snATAC-seq 峰显著增加。值得注意的是,Gpnmb之是脂肪肝的生物标志物。与RNA -seq结果一致,这种表观遗传变化伴随着Gpnmb基因表达的上调(图S9d-f)。然而,这种上调仅限于聚类2 和 3,它们在母体肥胖条件下按比例扩大。为了研究这些不同的 KC 状态是否在转录水平上受到差异调控,他们进行了 DEG 分析,并结合了 DecoupleR 转录因子活性分析。这表明 KC 状态按饮食条件而不是身份聚类,这表明转录因子活性的持续变化是驱动 HFDMCDLCD KC 中观察到的 DEG 的因素(图4c)。转录因子活性分析确定 HIF1α 在 HFDMCDLCD 条件下主要在聚类 1、2 和 3 中活跃。Jun、Ets1 和 Esr2 等转录因子在大多数 HFDMCDLCD 聚类中活性更高,其中 PPARα 和 PPARγ 在所有 HFD M CDL CD 聚类中均表现出活性增强(图4c)。这些发现表明,除了 HIF1α 之外,肥胖母鼠所生后代中其他持续的转录变化也源于转录因子活性的改变。
接下来,他们利用 snRNA-seq 数据通过 CellChat 分析评估了 HFDMCDLCD KC 中激活的信号通路。与RNA-seq 数据一致,在母体肥胖条件下,KC通过 APOE 和 APOA1 信号通路表现出细胞间通讯增加(图S9g)。基因表达分析证实了所有 HFDMCDLCD 状态下Apoe和Apoa1的上调(图4d),这与已知的载脂蛋白44调节因子 PPARα 和 PPARγ 的转录因子活性增加一致。此外,ATAC-seq 分析显示,与含有 PPARα 和 PPARγ-RXRα 转录因子结合侧的 CDMCDLCD 相比, HFDMCDLCD 聚类 3 中位于Apoe启动子上游的假定调控元件的峰值增加(图4e)。
为了研究载脂蛋白是否导致肝脏脂质蓄积,他们采用了肝细胞活体成像细胞培养方法,在培养基中添加了APOE或APOA1。他们使用肿瘤坏死因子(TNF)作为阳性对照,该因子此前已证实是KC衍生的肝细胞脂质蓄积驱动因子。在所有条件下,LD540信号强度均增加,其中APOE诱导的脂质蓄积量最高,与对照肝细胞相比(图4f)。
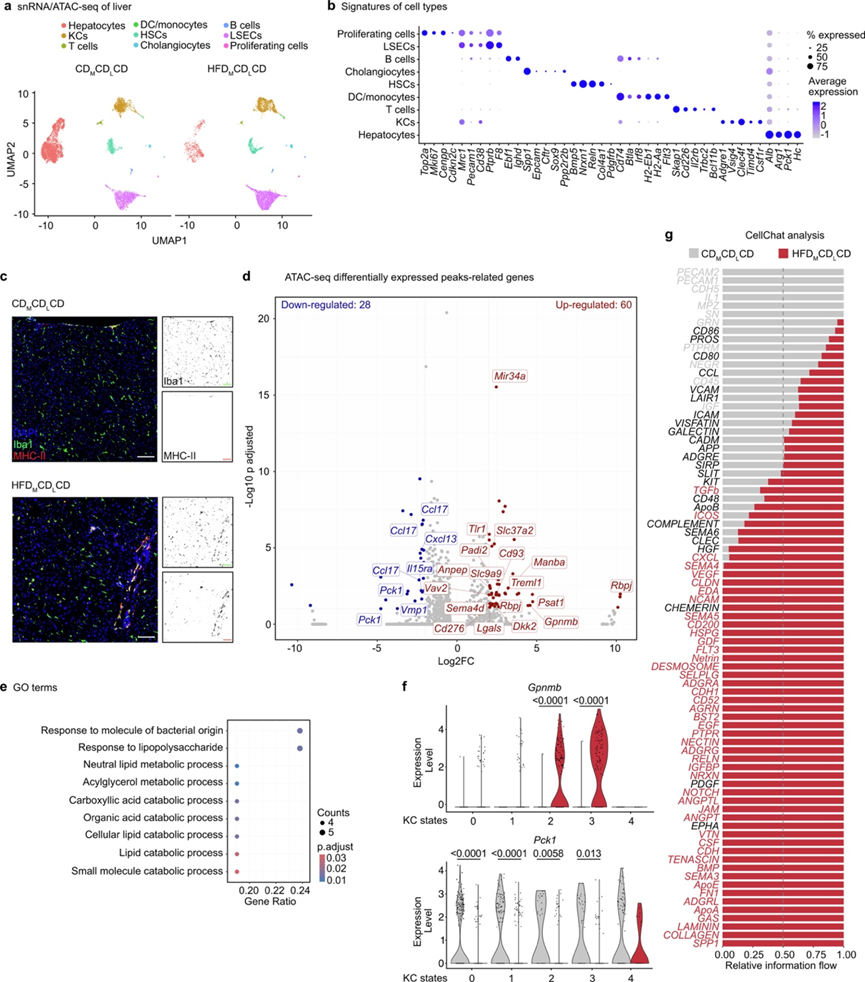
图S9. snRNA-seq和snATAC-seq。
(a) UMAP可视化。(b) 点阵图显示了决定细胞类型的基因。(c) 免疫荧光分析。(d) 火山图显示分配给 CDMCDLCD 和 HFDMCDLCD 肝脏的 KC 之间差异检测到的 snATAC-seq 峰的基因。(e)GO分析。(f) 所有KC状态下Gpnmb(上)和Pck1 (下)的表达。(g) CellChat分析。
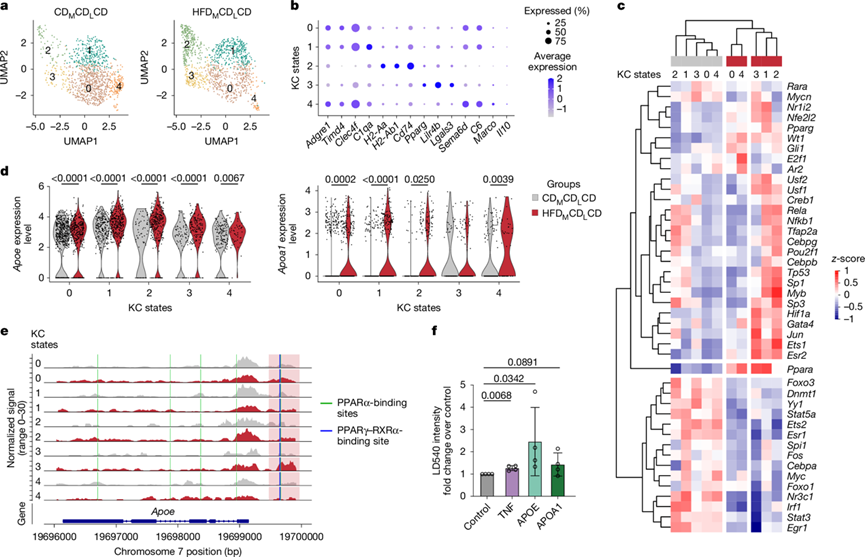
图4. 母鼠肥胖会诱发后代 KC 的表观遗传和转录变化。
(a) 五种不同 KC 状态的聚类分析。(b) 点图显示每个 KC 状态特异表达的基因。(c) 热图显示 CDMCDLCD 和 HFDMCDL CD 组每个聚类中预测的转录因子活性。(d) 所有 KC 状态下的Apoe(左)和Apoa1(右)的表达。(e) Apoe基因座的 ATAC-seq 峰覆盖图。(f) 在体外培养的肝细胞中,脂质积累情况以标准化LD540强度显示。
+ + + + + + + + + + +
结 论
本研究利用母体肥胖小鼠模型来扰乱妊娠期间的 KC 功能,肥胖母鼠的后代会患上脂肪肝疾病,这是由持续到成年的 KC 异常发育编程驱动的。程序化的 KC 通过载脂蛋白分泌促进肝细胞对脂质的吸收。肥胖母鼠所生新生小鼠的KC缺失,随后补充幼稚单核细胞,可挽救脂肪肝疾病。此外,在妊娠期间敲除巨噬细胞中编码HIF1α的基因,可阻止KC的代谢程序从氧化磷酸化转向糖酵解,从而避免脂肪肝疾病的发展。这些结果证实了KC功能的发育紊乱是成年期脂肪肝疾病的致病因素,并将胎儿来源的巨噬细胞定位为健康和疾病发育起源概念中关键的代际信使。
+ + + + +



