

 English
English文献解读|Cell Rep Med(10.6):对胃癌生态型进行15层多组学分析,可为治疗提供新的见解
✦ +
+
论文ID
原名:A 15-layer multi-omics analysis of gastric cancer ecotypes provides therapeutic insights
译名:对胃癌生态型进行15层多组学分析,可为治疗提供新的见解
期刊:Cell Reports Medicine
影响因子:10.6
发表时间:2026.04.20
DOI号:10.1016/j.xcrm.2026.102756
背 景
胃癌(GC)仍然是全球癌症相关死亡的主要原因之一,其预后不良主要归因于显著的生物学异质性和有限的治疗效果。包括癌症基因组图谱(TCGA)和亚洲癌症研究组(ACRG)在内的大规模基因组学和转录组学研究已经鉴定出关键驱动突变,并基于诸如EB病毒(EBV)感染、微卫星不稳定性(MSI)、染色体不稳定性(CIN)以及TP53活性和上皮间质转化(EMT)的转录特征等特征定义了分子亚型。然而,这些研究未能完全阐明下游功能后果,并且依赖于整体测量,而这掩盖了细胞异质性和肿瘤微环境(TME)结构。
实验设计
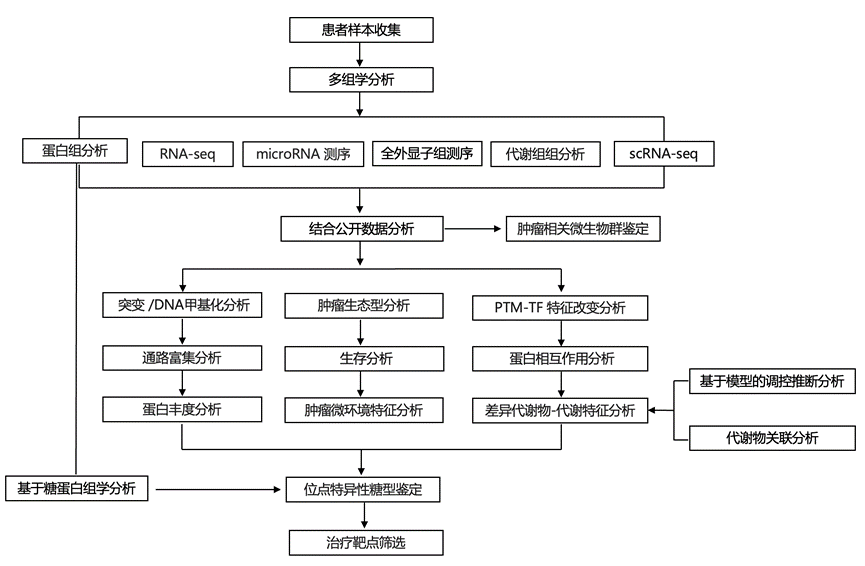
结 果
01

CPTAC GC 队列的多组学分析
为了全面表征胃癌的异质性,研究团队构建了一个包含165例未经治疗的原发性非贲门胃腺癌和41例配对的、经组织学证实的正常邻近组织(NAT)的队列,排除了已知具有遗传性癌症易感性的患者。最终保留了159例肿瘤和30例NAT。该CPTAC队列反映了胃癌在年龄、临床记录的性别、组织学类型和分期方面的多样性(图1 A),并且主要来自东欧患者,是对先前以亚洲人群为主的蛋白质组学研究的补充。
将均质化的速冻组织进行多组学分析。全外显子组测序和全基因组测序(WGS)用于鉴定体细胞突变和拷贝数变异(CNA);甲基化芯片用于检测DNA甲基化;转录组分析(RNA-seq)和microRNA测序用于定量mRNA、环状RNA和miRNA的丰度;串联质谱标签蛋白质组学用于测量蛋白质的整体丰度、磷酸化、糖基化和乙酰化水平;数据非依赖性采集蛋白质组学用于定量泛素化;交联质谱(XL-MS)用于绘制蛋白质-蛋白质相互作用(PPI)图谱;代谢组学用于分析肿瘤代谢物;基于WGS和RNA-seq数据推断肿瘤相关微生物群;并从元数据中提取临床注释。这些分析共生成15个数据层,包含超过385000个高置信度特征(图1B)。他们进一步推导出了更高阶的特征,包括基因组畸变指标、肿瘤组成指标、调控和通路活性评分、TCGA 4和 ACRG 5亚型分类以及微生物群落概况(图 1 C)。
在157例基因组分析的肿瘤中,TCGA分类识别出EBV阳性(8%)、微卫星不稳定性(MSI)(19%)、染色体不稳定性(CIN)(35%)和基因组稳定(GS;38%)亚型。在微卫星稳定(MSS)肿瘤中,复发性突变与之前的报道一致,包括TP53、ARID1A、PIK3CA、CDH1、MAP2K7和SMAD4(图1D)。MSI肿瘤的突变频率高于TCGA报道(图1E),这可能反映了测序深度的增加。体细胞 CNA 分析发现了臂水平和局灶性改变,包括 17q12 (ERBB2)、20q13.2 (AURKA)、8q24.21 (MYC)、6p21.1 (VEGFA) 和 19q12 (CCNE1) 的复发性局灶性扩增(图 1 F)。
深度PTM分析显著扩展了已知的PTM图谱,超越了现有数据库和CPTAC3研究的范围。在本研究中鉴定的PTM位点中,6.1%的丝氨酸/苏氨酸/酪氨酸(S/T/Y)磷酸化位点、5.3%的赖氨酸(K)乙酰化位点、23.8%的赖氨酸泛素化位点和17.9%的天冬酰胺(N)-连接糖基化位点均为新发现(图1G)。该PTM图谱为机制研究和治疗发现提供了更多机会。例如,JAK2是一种受泛素介导的周转调控的关键免疫调节激酶,本研究发现了21个此前未注释的泛素化位点和1个新的磷酸化位点(图1H)。此外,他们在TACSTD2(TROP2)的治疗表位处或附近检测到了糖基化和泛素化位点(图1I),TACSTD2是美国食品药品监督管理局 (FDA) 批准的抗体药物偶联物 (ADC) 靶向的表面抗原,目前正在进行GC的临床评估,这凸显了将PTM信息纳入治疗表位选择和生物标志物开发的重要性。
他们在肿瘤队列中建立了大规模的XL-MS分析,构建了一个GC特异性的PPI网络,该网络显著扩展了已收录的PPI数据库(图1J)。凭借残基水平的分辨率,该网络揭示了新的相互作用界面,并为研究不足的蛋白质提供了背景信息。例如,特征不明确的癌睾丸抗原CCDC168与EMT相关蛋白VIM和TPM2相互作用,同时还证实了已知的VIM相互作用(图1K)。
定量整合揭示了广泛的翻译后调控。mRNA-蛋白质相关性中位数为中等,与其他癌症一致,而PTM-蛋白质相关性同样较低,PTM-mRNA相关性则较弱(图1 L)。
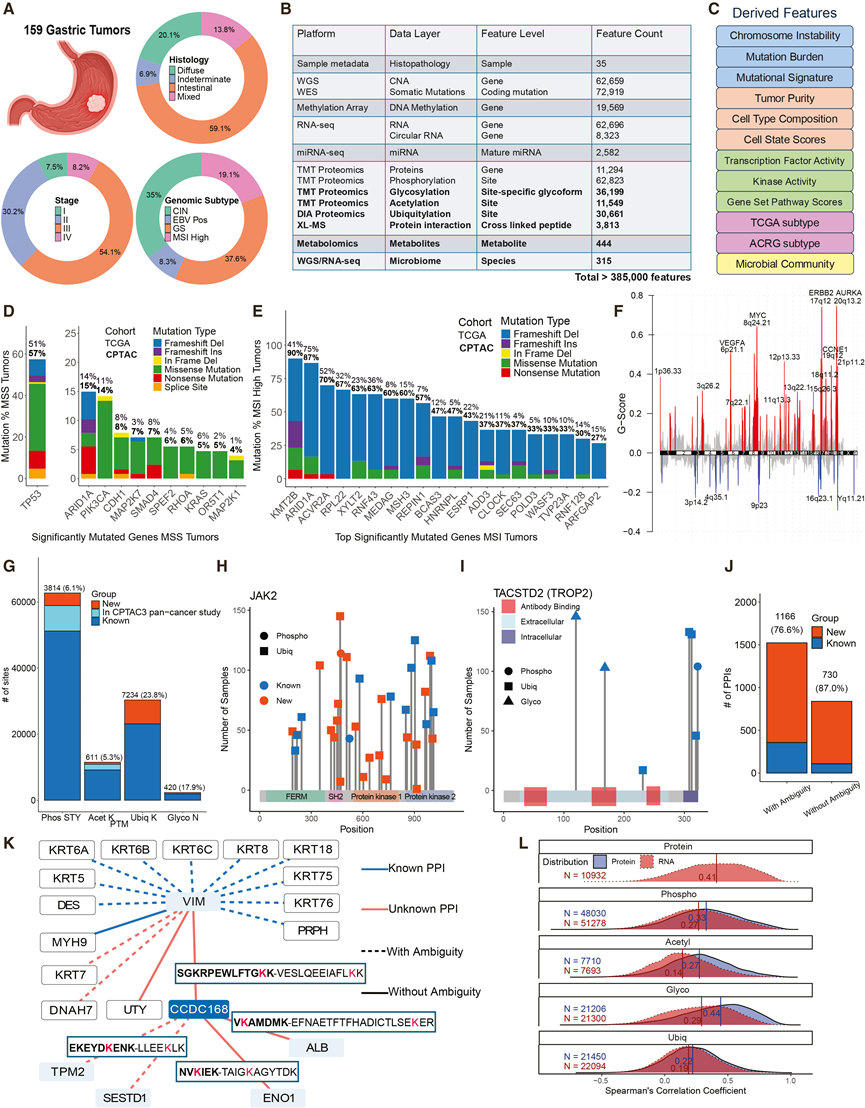
图1. CPTAC GC 队列的多组学图谱。
(A) 队列中临床特征的分布。(B) 15 个多组学数据层的概述。(C) 从(B)中提取的特征,按类别分组和着色:基因组异常指标、肿瘤组成指标、调控活性、分子分类和微生物群落。(D) MSS 肿瘤中显著突变基因的突变频率。(E) MSI-H 肿瘤中前 20 个显著突变基因的突变频率。(F) G 分数反映改变频率和幅度。(G) 本研究中鉴定的 PTM 位点。(H) JAK2 上鉴定的 PTM 位点的棒棒糖图。(I) JAK2 上鉴定的 PTM 位点的棒棒糖图。(J) 通过 XL-MS 鉴定的 PPI,在 STRING、BioGRID、BioPlex 或 HuRI 中归类为新鉴定的相互作用或已知相互作用。(K) XL-MS 鉴定了 CCDC168 及其伴侣蛋白 VIM 的蛋白质-蛋白质相互作用 (PPI)。(L) 肿瘤中Spearman相关性的分布。
02
基因组和表观基因组异常的功能背景分析
他们评估了MSS肿瘤中11个显著突变基因的突变状态与转录组、蛋白质组、PTM和代谢组等多个层面特征之间的关联(图1D)。TP53、MAP2K1和SMAD4显示出最广泛的分子特征(图2A)。TP53突变与广泛的转录和磷酸化信号通路改变相关,这与TP53作为基因表达和应激信号通路主调控因子的作用相符。经典的TP53诱导基因MDM2、DDB2和RPS27L的mRNA关联性最强(图2A)。MAP2K1突变与广泛的转录组改变相关,尽管可检测到的磷酸化位点改变极少,这表明触发下游转录反应的磷酸化事件可能较为狭窄或短暂。相比之下,SMAD4突变与广泛的转录后重塑相关,包括广泛的蛋白质、翻译后修饰和代谢物变化,以及激酶活性改变(图2B)。激酶活性改变由激酶底物磷酸化水平的改变所支持,且该改变与总蛋白丰度无关。例如,在SMAD4突变肿瘤中,多个CSNK2A1底物的磷酸化水平升高,而mRNA或蛋白水平并未发生相应变化(图2A-B)。虽然SMAD4敲低与CSNK2A1表达增加相关,SMAD4突变肿瘤中CSNK2A1激酶活性增强,提示其可能是一个潜在的治疗靶点。
接下来,他们优先筛选出拷贝数、mRNA 和蛋白质改变一致的CNA,以及肿瘤和NAT之间蛋白质丰度差异显著的CNA,从而鉴定出 1729 个候选驱动基因,这些基因富集于 RNA 加工、核转运和氨基酸代谢(图2C-D)。其中,在肿瘤中频繁上调的扩增 RNA 剪接因子尤为突出(图 2E)。这些因子与MYC表现出很强的mRNA相关性(图2F),表明在MYC扩增的肿瘤中存在协同上调,并提示可能存在癌基因诱导的依赖性。
通过对DNA甲基化的评估,他们鉴定出102个候选甲基化驱动基因,这些基因与mRNA和蛋白质表达呈负相关,且富集于细胞间连接组织和神经肽信号通路(图2G-H)。在这些与连接相关的基因中(图2I),CLDN18具有治疗意义,因为其亚型CLDN18.2是FDA批准的胃癌抗体疗法的靶点。甲基化探针定位于CLDN18.1,该基因在肿瘤中甲基化水平升高,mRNA表达降低,表明CLDN18基因位点存在表观遗传抑制。同时,与正常组织相比,肿瘤中CLDN18.2的mRNA和蛋白质水平也较低(图2J)。值得注意的是,肿瘤中上皮标志物CDH1的蛋白水平显著降低(图2J),这与EMT相关的上皮连接破坏相一致。这些结果共同表明,CLDN18.2的治疗意义可能反映的是EMT过程中膜可及性的改变,而非肿瘤特异性过表达,这与之前的报道一致。由于CLDN18.2高表达的肿瘤同时具有上皮和间质状态(图2K),因此,结合其他标志物,例如CLDN18.1甲基化、CLDN18 mRNA水平或EMT指标,可能有助于更好地识别正在经历EMT的肿瘤,同时保留可及的CLDN18.2以进行靶向治疗。
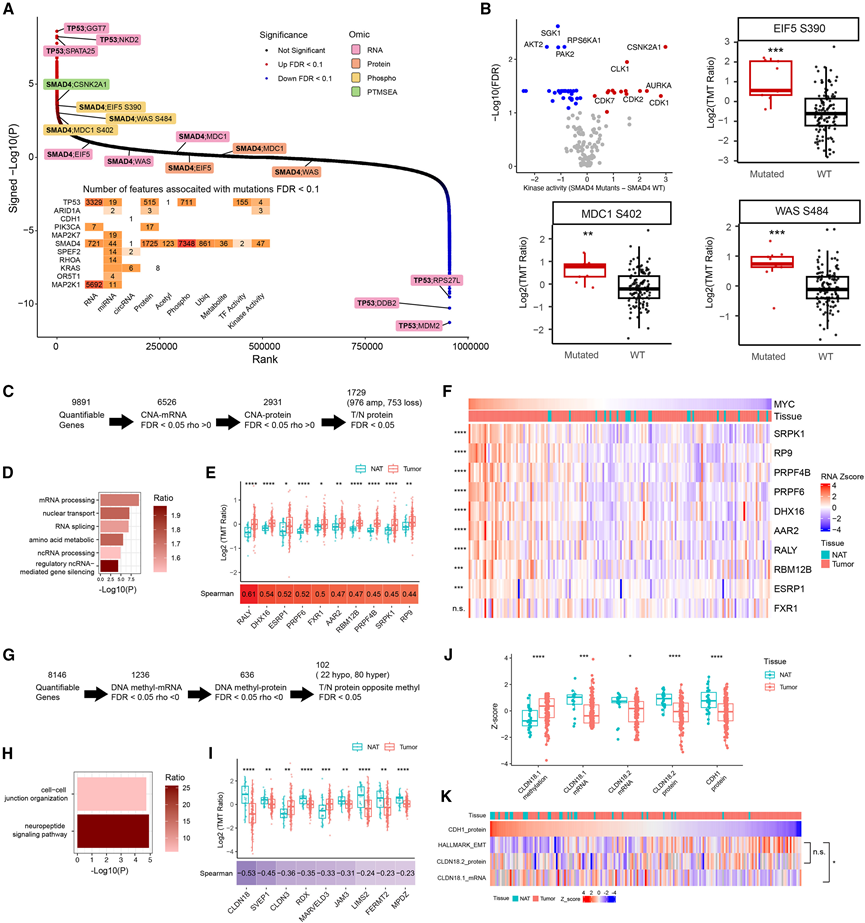
图2. 基因组和表观基因组异常的功能背景。
(A) MSS 肿瘤中基因特征关联。(B) SMAD4突变体和SMAD4野生型 MSS 肿瘤之间激酶活性差异的火山图。(C) 通过整合 CNA-mRNA 和 CNA-蛋白质 Spearman 相关性与肿瘤与 NAT 差异表达(WRS)来确定候选 CNA 驱动因素优先级的工作流程。(D) GO分析。(E) 肿瘤与NAT中10个优先考虑的RNA剪接相关基因的蛋白质丰度箱线图。(F) MYC 和(E)中剪接相关基因的mRNA水平热图。(G) 通过整合甲基化-mRNA和甲基化-蛋白质Spearman相关性与肿瘤与NAT差异表达(WRS)来确定候选甲基化驱动因素优先级的工作流程。(H)GO分析。(I) 肿瘤与NAT中参与细胞间连接组织的九个基因的蛋白质丰度箱线图。(J) 箱线图比较肿瘤和NAT的CLDN18.1甲基化和mRNA、CLDN18.2 mRNA和蛋白质以及CDH1和VIM蛋白质表达。(K) 箱线图比较肿瘤和NAT的CLDN18.1甲基化和mRNA、CLDN18.2 mRNA和蛋白质以及CDH1和VIM蛋白质表达。
03
肿瘤生态型揭示了超越基因组亚型的TME驱动分层
为了超越以基因组为中心的分类并更好地解析TME结构,他们应用了一种经过验证的RNA-seq反卷积方法来评估71种上皮细胞、间质细胞和免疫细胞状态的相对水平。共识聚类分析识别出六种肿瘤生态型(C1-C6)(图3A)。对37种显著变异的细胞状态进行聚类分析,定义了五种共存的TME程序(图3A):过渡界面程序,反映了上皮-间质细胞间的相互作用和低度炎症;免疫效应细胞富集程序,代表炎症和免疫激活的TME;初始启动免疫程序,表明免疫浸润但激活有限;免疫调节程序,反映了免疫排斥的、维持组织的微环境,可能有助于肿瘤的持续存在和进展;以及血管生成免疫抑制程序,代表促肿瘤发生的TME。在五个具有可用生存数据的外部GC队列(来自四项独立研究)中,Cox回归和meta分析表明,过渡界面和免疫效应富集程序与良好的预后相关,初始启动免疫是中性的,而免疫调节和血管生成免疫抑制程序预测不良结果(图 3A)。
不同的生态型代表了TME程序的不同组合(图3A)。C1富集免疫调节和血管生成免疫抑制程序,具有最高的基质含量(图3B),并富集ACRG EMT亚型和弥漫性组织学特征。C2以血管生成免疫抑制为主,不伴有免疫调节特征。C3由初始免疫程序定义。C4符合过渡界面程序,具有最高的肿瘤纯度和上皮含量(图3B)。C5结合了免疫效应富集和初始免疫程序,显示出最高的免疫浸润(图3B),表达CD274(PD-L1)和IDO1,富集MSI相关突变特征SBS15,且TP53突变较少。C6是出现频率最低的生态型,其AGR2+上皮细胞选择性富集,提示存在一种独特的、以上皮细胞为中心的TME。在临床特征中,仅年龄与生态型相关,C4型患者的年龄高于C1型和C3型患者。
利用本研究队列中的生态型标签和细胞状态谱训练的多分类器,在五个具有不同祖先和地理组成的外部胃癌数据集上重现了生态型模式。这些外部数据集的生存分析显示,C1预后最差,C2和C3预后中等,C4–C6预后良好(图3 C)。多变量Cox回归分析,校正年龄、临床记录的性别、分期和Lauren亚型后,证实生态型是独立的预测因子。虽然C1和C2有血管生成免疫抑制程序,但C1的预后比C2更差,这表明免疫调节和组织维持程序的额外存在可能导致更具抑制性和治疗抵抗性的TME。由于本研究队列缺乏足够的长期生存数据,他们基于TCGA和ACRG数据集训练了一个扩散增强型人工智能模型,用于预测生存期。将该模型应用于本研究的队列后,该模型独立识别出C1是预后最差的生态型。
在本研究的队列和TCGA/ACRG队列中,根据TME结构,生态型将基因组相似的肿瘤进行了分层(图3D-E)。GS肿瘤富集于C1和C3型,而CIN肿瘤富集于C2和C4型。分类为C1和C3的GS肿瘤在TME模式(图3A)和生存结局(图3F)方面存在显著差异。弥漫型肿瘤也同样分为C1和C3型(图3G-H),且预后不同(图3I)。微卫星不稳定性(MSI)是免疫检查点阻断(ICB)的公认生物标志物。有趣的是,虽然MSI肿瘤主要为C5型,但队列中45%的MSI肿瘤和TCGA/ACRG队列中47%的MSI肿瘤属于其他生态型(图3D-E)。使用PredictIO 泛癌基因表达特征来预测 ICB 反应,C5 MSI 肿瘤的预测反应明显高于其他 MSI 肿瘤(图3J),这表明生态型可以进一步细化免疫疗法的分层,而不仅仅局限于 MSI。
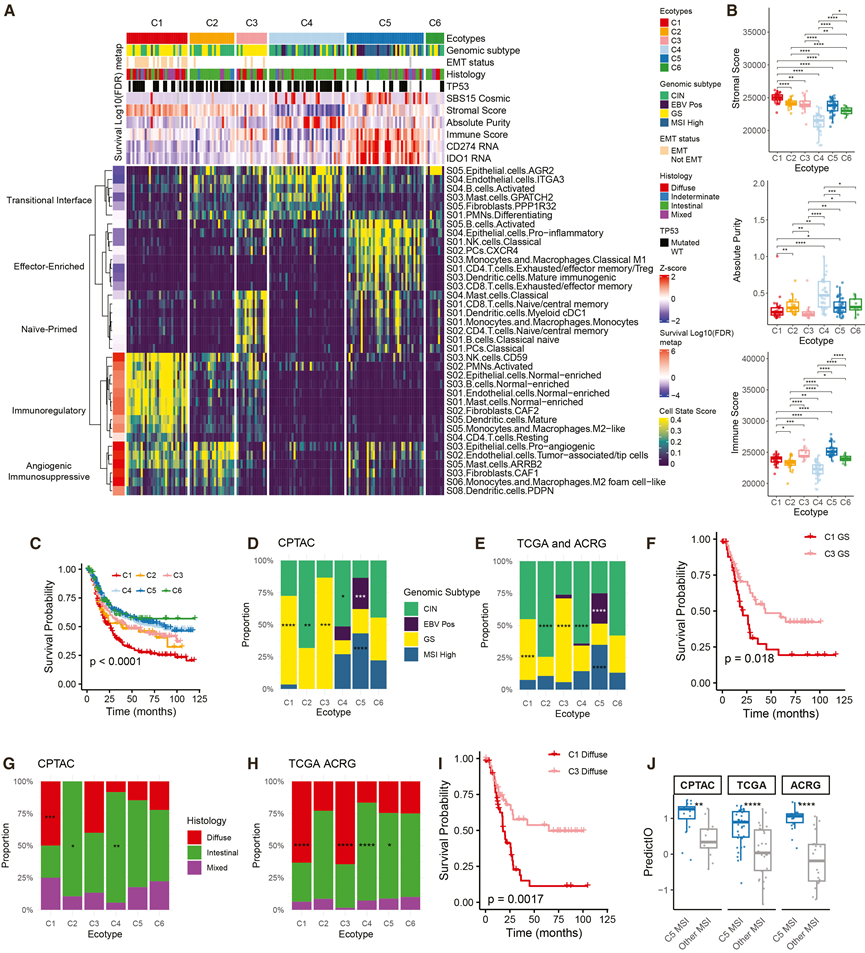
图3. TME 定义的肿瘤生态型对基因组和组织学亚型进行分层。
(A) 通过细胞状态评分的共识聚类鉴定出六种肿瘤生态型。(B) 箱线图比较不同生态型的纯度、免疫和基质评分。(C) 使用汇总的 TCGA、ACRG、GSE15459、GSE26899 和 GSE26901 数据对肿瘤生态型进行 Kaplan-Meier 生存分析。(D-E) 堆叠条形图显示了 CPTAC和 TCGA/AGRG队列中按生态型划分的基因组亚型分布。(F) Kaplan-Meier 生存分析。(G-H) 堆叠条形图显示 CPTAC 队列和 TCGA/AGRG 队列中按生态型划分的组织学亚型分布。(I) Kaplan-Meier 生存分析。(J) 在 CPTAC、TCGA 和 ACRG 队列中,C5 MSI 与其他 MSI 肿瘤的 PredictIO 免疫检查点反应评分。
04
蛋白质基因组程序是肿瘤生态型的基础
为了确定 GC 生态型背后的分子程序,他们整合了转录特征信号;转录因子 (TF)、激酶和 miRNA 的推断活性;多层PTM分析;以及基于XL-MS的蛋白质相互作用图谱。标志性分析揭示了不同的生态型特异性程序(图 4A)。C1富集了基质通路,包括EMT、血管生成和缺氧,这与其富含基质的表型以及ACRG EMT 亚型关联相一致。C4以增殖和生物合成特征为主,反映了其高度增殖的上皮状态。C5 表现出广泛的免疫相关程序激活以及代谢通路的抑制,支持其免疫炎症表型。
PTM 分析揭示了大量 mRNA 或蛋白质水平无法捕捉到的生态型特异性调控事件,包括4818 个磷酸化位点、259个乙酰化位点、631 个泛素化位点和1587 个完整的糖肽(图 4E)。在已注释的 PTM 位点中,许多位点的变化与已知的功能后果相符。例如,在 C1 中,DEAF1(T432)和PAK2(S197) 的激活性磷酸化水平升高与推测的转录因子和激酶活性增强平行;IDH2 (K106) 的抑制性乙酰化水平升高提示酶活性受到翻译后抑制;ANXA1 (K312) 的降解性泛素化水平降低与蛋白质丰度升高相对应(图4F-I)。相比之下,大多数生态型相关的 PTM 事件的功能仍未明确,代表着一个尚未充分探索的调控层面。例如,虽然LRP1 蛋白丰度降低,但 C5 中位点特异性糖型 LRP1-N1575 N4H5F0S1G0 升高(图 4 J)。
XL-MS 分析鉴定出数百个与生态型、EMT 状态或肿瘤-NAT 差异相关的显著调控的蛋白质内或蛋白质间相互作用。虽然大多数相互作用的变化与蛋白质丰度相关,但有些相互作用的发生与蛋白质表达无关。例如,EEF2 中的一个蛋白质内交联在 C5 中升高,蛋白质水平稳定(图4K)。接下来,他们使用 XL-Ranker 构建了一个 GC 特异性蛋白质相互作用网络,以解决肽映射的歧义,该网络包含 1248 个蛋白质编码基因,这些基因通过1185个高置信度的相互作用连接。使用 NetSAM进行分层模块分析,鉴定出102个网络模块,这些模块分为四个层级(图4L),涵盖代谢、免疫、基因调控以及细胞骨架和细胞结构。模块水平差异分析揭示了与生态型、EMT 和肿瘤 NAT 相关的改变,其中 C1 和 EMT 肿瘤共有多个受影响的模块。例如,染色质相关模块(L2M9)在肿瘤中表达上调,并按生态型分层,在预后良好的生态型(C4-C6)中表达最高,在预后不良的C1中表达最低(图 4 M)。相反,细胞骨架模块(L1M6)在肿瘤中普遍降低(图4 M),但在 C1 中相对保留,这与其维持组织微环境 (TME) 的特征相符。免疫相关模块(L2M7)在肿瘤中普遍上调,在免疫浸润的 C5 中上调最为显著(图4M)。虽然已知 L2M7 中的几个基因在免疫细胞中表达,但该模块内的所有相互作用都是通过 XL-MS 新发现的,这突显了能够揭示未经整理的数据库之外的网络背景的能力。
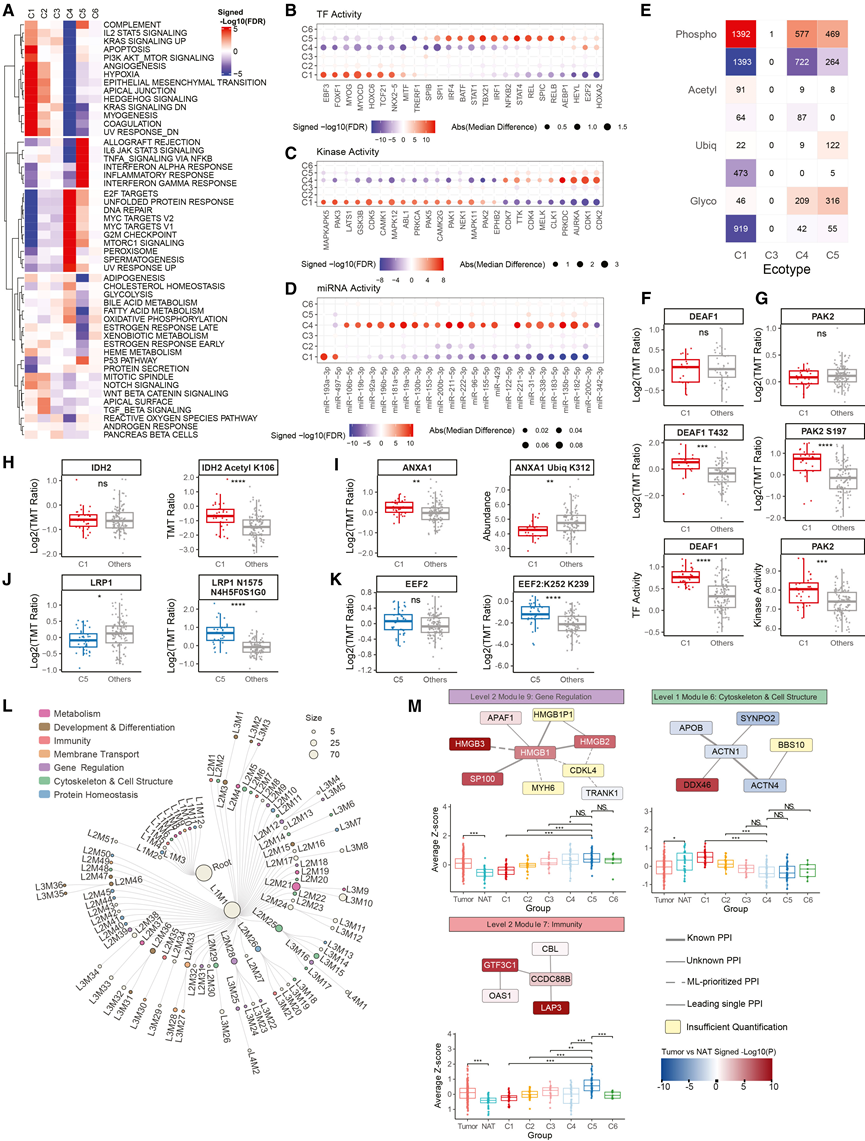
图4. 蛋白质基因组程序支撑肿瘤生态型。
(A) 不同生态型蛋白质组学衍生Hallmark通路活性的热图。(B-D) 在单样本与WRS中,差异活性最强的25个转录因子、激酶和miRNA。(E) 通过单与WRS按 PTM 类型显著调控的 PTM 位点的热图。(F-J) 箱线图比较特定生态型与其他生态型之间的蛋白质、PTM 或推断活性。(K) C5 与其他组相比,EEF2 蛋白丰度和蛋白内交联 K252-K239 的箱线图。(L) 源自 XL-MS PPI 网络的模块的层次结构。(M) 来自(L)的代表性模块,显示了不同生态型以及肿瘤与NAT之间的差异丰度。
05
代谢程序是肿瘤生态型的基础
为了确定与肿瘤生态型、EMT状态和肿瘤-NAT差异相关的代谢程序,他们整合了代谢组学、转录组学、蛋白质组学和PTM数据。比较分析鉴定出220种与肿瘤生态型、EMT状态和肿瘤-NAT差异相关的代谢物(图5A)。在C5型肿瘤中,喹啉酸和犬尿氨酸水平升高,这与免疫浸润肿瘤中免疫抑制性色氨酸-犬尿氨酸通路的激活相一致。C4型肿瘤显示胸苷和胸腺嘧啶水平升高,反映了增殖状态下核苷酸需求的增加。相反,C1型和EMT型肿瘤中甘油-3-磷酸水平升高,这与EMT过程中脂质代谢的重编程相一致。
在84种具有可量化前体的代谢物中,每种代谢物的最大前体-代谢物相关性的中位数Spearman相关系数为0.43(图5B),表明除了底物可用性之外,还存在显著的酶促调控。为了系统地鉴定除前体效应之外的候选调控因子,他们应用了MetaSage,这是一个机器学习框架,它通过整合前体水平以及直接相关的合成代谢和分解代谢酶的mRNA、蛋白质和PTM测量值来模拟每种代谢物的丰度(图5C-E)。在220种差异代谢物中,114种具有可量化的预测因子,其中51种的预测结果稳健。对于25种代谢物,模型预测值与实测丰度的相关性强于任何单个前体(图5F),表明酶促调控发挥了作用。特征重要性分析构建了一个调控网络,将196个重要特征与51种预测良好的代谢物联系起来(图S5B;STAR方法)。前体代谢物占这些预测因子的26%,相关酶的RNA和蛋白质表达各占22%,翻译后修饰占30%(图5G)。
IDO1 是甲酰犬尿氨酸丰度的最佳预测因子,这与其催化色氨酸转化为甲酰犬尿氨酸的作用相符(图 5H)。IDO1 mRNA 和蛋白在C5生态型中均升高(图 3A、图5I-J),与甲酰犬尿氨酸增加和色氨酸减少相一致(图5C)。支持这一结论的是,介导下游甲酰犬尿氨酸代谢的 AFMID 在C5中降低。scRNA-seq数据显示IDO1在树突状细胞中富集,表明TME)参与调控色氨酸代谢。
IDO1 K389的泛素化与甲酰犬尿氨酸水平相关(图5E),并在C5中升高(图5K),与 IDO1 mRNA和蛋白丰度呈正相关(图5L),表明K389的泛素化在此情况下并不促进降解。结构定位显示K389位于C端K螺旋内,由周围的螺旋和 EF 环稳定(图5M),这可能限制了其可及性。先前的研究表明,该区域的结构暴露可导致K389泛素化介导的降解,这支持了其作为条件性降解子和胃癌潜在治疗靶点的作用。
最后,将代谢紊乱物与五种TME程序进行关联分析(图3A),鉴定出程序特异性的代谢特征(图5N)。甲酰犬尿氨酸、犬尿氨酸和喹啉酸与免疫效应富集程序密切相关,强化了免疫活性TME中IDO1介导的色氨酸代谢。相反,包括果糖、半乳糖、葡萄糖和甘露糖在内的糖酵解途径单糖与免疫调节程序相关,提示其适应低氧、富含基质的微环境。通路富集分析进一步支持了这些关联(图5O)。
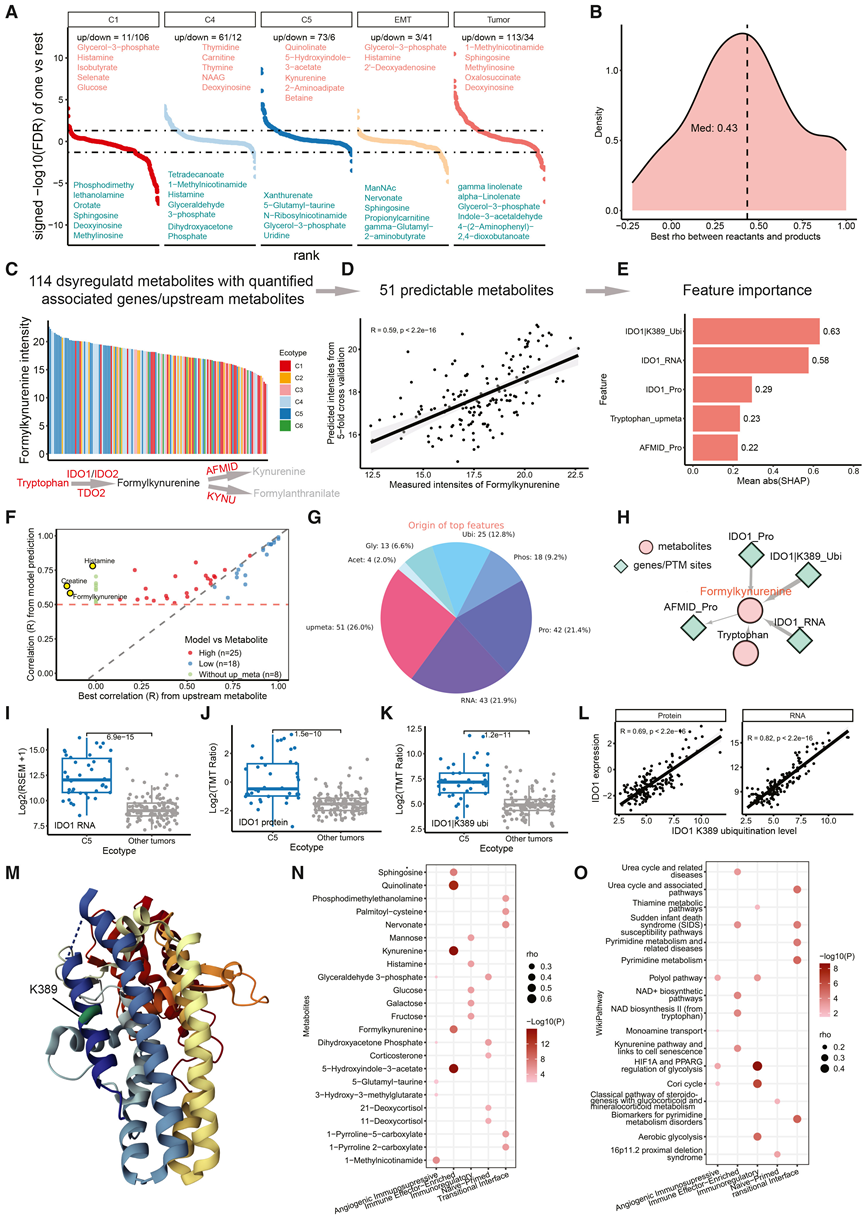
图5. 代谢失调的整合分析及基于模型的调控推断。
(A)鉴定出的差异表达代谢物。(B) 每个显著变化的代谢物与其定量上游反应物之间最强Spearman相关性的分布。(C) 不同生态型中甲酰犬尿氨酸的强度。(D) 甲酰犬尿氨酸强度与模型预测的甲酰犬尿氨酸强度之间的Spearman相关性。(E) SHAP(Shapley Additive Explanations)特征重要性得分。(F) 散点图比较每种代谢物的模型预测丰度与实测丰度之间的Pearson相关系数(y轴)以及实测丰度与任何已定量上游代谢物之间最强的相关性(x轴)。(G) 顶级预测特征的来源:上游反应物(红色)和基因衍生特征(蓝色),不同的蓝色色调表示不同的基于基因的特征组。(H) 以甲酰犬尿氨酸为中心的网络,显示其主要预测特征。(I-K) C5 与其他生态型中 IDO1 RNA、蛋白质和 K389 泛素化的丰度。(L) IDO1 K389 泛素化与 IDO1 RNA/蛋白质表达之间的 Spearman 相关性。(M) IDO1 上 K389 的结构位置。(N-O) 基于 Spearman 相关性,针对每个细胞状态群落,鉴定出前五种正相关的代谢物和富集的代谢通路。
06
肿瘤微生物组与肿瘤亚型和分子程序的关联
他们利用经过全面且严格筛选的微生物分析流程处理的WGS和RNA-seq数据,对肿瘤相关微生物群进行了分析,鉴定出多种细菌、真菌和病毒(图6A)。在最初检测到的 1506 个物种中,去除可能的污染物和假阳性结果后,最终得到315个高置信度、组织富集的物种。在所有肿瘤样本中,链球菌属和普雷沃氏菌属最为丰富,其次是其他胃肠道相关菌群,例如乳杆菌属、梭杆菌属和螺杆菌属。EBV 在 13 个样本中富集(图6A),与独立确定的 EBV 亚型完全匹配(每个肿瘤细胞约10.6个病毒拷贝)。
对肿瘤微生物组的群落分析鉴定出一个仅由EB病毒(EBV)组成的菌群,以及另外四个包含多种微生物的菌群,分别命名为幽门螺杆菌(Helicobacter pylori)、乳杆菌-白色念珠菌(Lactobacillus-Candida albicans)、致病有机体(Pathobiont)和韦荣氏球菌(Veillonella parvula)(图6B)。其中,致病有机体最为丰富。总体而言,肿瘤可分为五种“肿瘤肠型”,这些肠型主要由特定的微生物菌群组成(图6C)。
在27对匹配的肿瘤-NAT样本中,幽门螺杆菌和韦荣氏球菌在NAT样本中富集。在物种水平上,幽门螺杆菌在NAT样本中富集,而EB病毒在肿瘤中富集(图6 D),这支持了幽门螺杆菌促进GC发生而EB病毒促进肿瘤进展的模型。
不同肿瘤生态型的微生物组成也存在差异。C4 和 C5肿瘤的细菌负荷较高(图 6E),且富含致病有机体(图 6F)。物种水平分析显示,C4 肿瘤中S. anginosus和其他致病菌群物种富集,而 C5 肿瘤中则富集了加氏乳杆菌和Limosilactobacillus vaginalis(图 6G)。此外,S. anginosus与上皮间质转化(EMT)状态呈负相关。在MSI基因组亚型中,C4 肿瘤的致病菌群相对丰度高于 C5 肿瘤(图6H),提示低免疫浸润可能有利于致病菌的定植。
为了探索功能关联,他们首先评估了肿瘤糖基化谱,因为它们在宿主-微生物相互作用中发挥作用。富含病原菌聚类的肿瘤显示岩藻糖-唾液酸聚糖水平降低,而高甘露糖聚糖水平升高;后者与肿瘤增殖有关(图6I)。
整合代谢组学和蛋白质组学分析揭示了与病原菌群最广泛的关联(图6J)。基于蛋白质组学的通路富集分析将病原菌群与代谢重编程、RB信号通路、DNA错配修复、缺氧、NF-κB介导的炎症以及5-氟尿嘧啶(5-FU)化疗耐药性联系起来(图6K)。代谢物分析将病原菌群与组氨酸代谢联系起来(图6K),包括N-甲酰亚氨基谷氨酸(FIGLU)水平升高(图6L),FIGLU是叶酸缺乏的生物标志物。由于叶酸缺乏会导致5-FU耐药性,因此这种关联提示微生物与化疗耐药性之间存在联系。胸苷酸合成酶 (TYMS) 是 5-FU 耐药性的介质,也与病原菌聚类相关(图 6 M)。
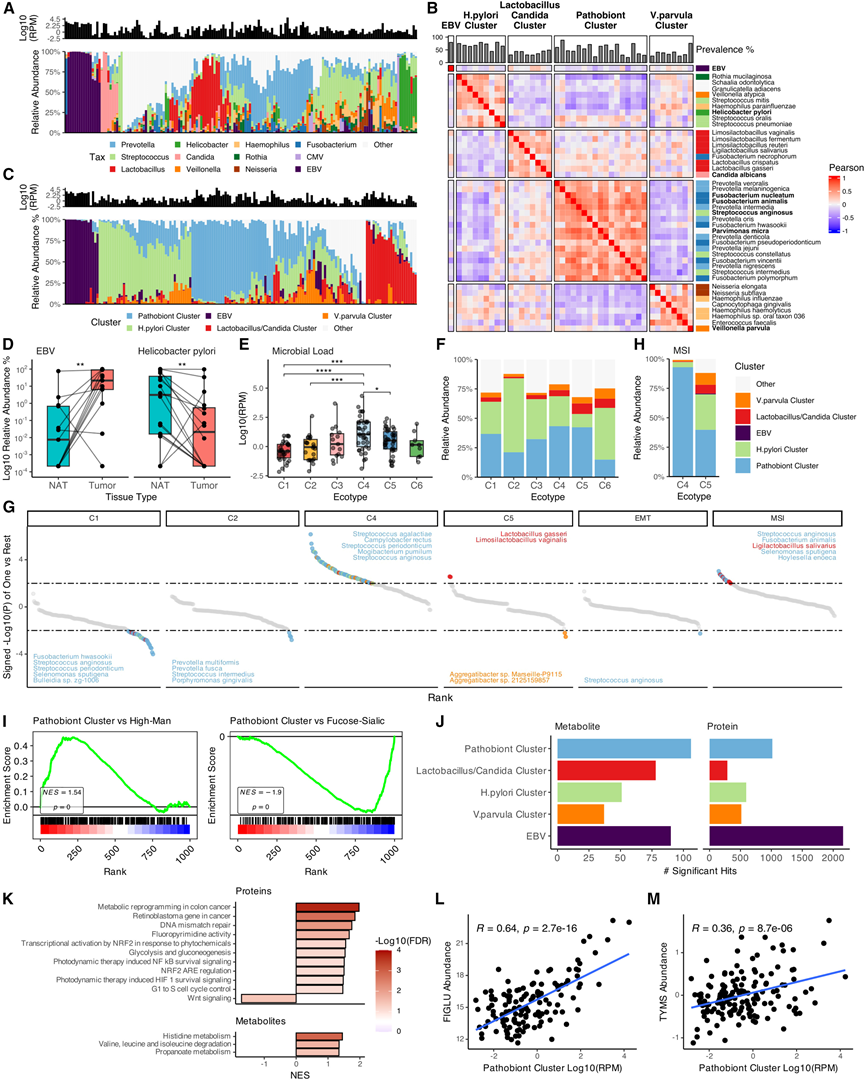
图6. 胃组织相关微生物组的特征及其与肿瘤特征的关联。
(A)全基因组测序原发肿瘤的微生物组成。(B) 物种丰度和微生物群落的Spearman相关性热图。(C) 微生物群落组成:微生物总负荷(上)和群落丰度(下)。(D) 配对肿瘤-正常组织分析显示肿瘤中 EBV 富集。(E) 按生态型划分的肿瘤微生物负荷。(F) 按生态型划分的微生物群落相对丰度中位数。(G) C4-MSI和 C5-MSI肿瘤中微生物聚类丰度中位数的相对丰度。(H) 利用单一物种与其余物种丰度分析,按生态型和亚型进行物种富集分析。(I) 山形图显示与病原体聚类相关的高甘露糖和岩藻糖-唾液酸聚糖类型的富集和减少。(J) 按生态型和亚型分组后计算的显著聚类-代谢物(上)和聚类-蛋白质(下)Pearson 相关性的数量。(K) 从与病原菌聚类丰度相关的蛋白质(上图)和代谢物(下图)中鉴定出的显著富集通路。(L) 病原菌聚类丰度和 FIGLU 丰度的散点图。(M) 病原菌聚类丰度与TYMS表达的散点图。
07
在GC异质性背景下进行治疗靶点优先排序
由于胃癌表现出广泛的基因组、分子和微环境异质性,传统的肿瘤-正常组织比较可能忽略亚组特异性靶点。因此,他们进行了系统的异常值分析,以识别在肿瘤亚群中异常激活的蛋白质和翻译后修饰,并根据已发表的scRNA-seq数据,将它们映射到基因组亚型、生态型和细胞起源上。
77%的肿瘤表现出至少一种已知的基于抗体和T细胞的靶点的异常高表达,这些靶点包括ERBB2(HER2)、TACSTD2(TROP2)、CEACAM5、MUC1和FOLR1(图7A)。他们还发现了其他具有反复高表达异常值的表面蛋白(图7A),包括COL12A1、COL5A1和VCAN,这些蛋白的mRNA水平与既往队列研究中的不良生存率相关,从而扩展了候选表面靶点的范围。
利用完整的糖蛋白组学数据,他们鉴定出具有反复高表达异常值的位点特异性糖型(图7B)。在具有≥15个高表达异常值的位点特异性糖型中,高甘露糖型显著富集(图7C),表明N-聚糖生物合成不完全,并可能与肿瘤中甘露糖去除酶MAN2C1以及岩藻糖基化和唾液酸化酶FUT1、FUT2、ST6GALNAC1和ST3CAL6的下调有关。在基因水平上,340个基因至少包含一个高表达的位点特异性糖型异常值。CEACAM5排名第四,70%的肿瘤显示出糖基化异常值,许多其他抗体药物靶点也显示出广泛的糖肽异常值(图7D)。值得注意的是,COL1A2 和 COL5A2 显示出适度的蛋白质异常值(12 和 14 个样本),但位点特异性糖型异常值却很广泛(图7E),这突显了聚糖重塑可以显著扩展和细化治疗靶点范围,而不仅仅取决于蛋白质丰度。
接下来,他们利用 DepMap中来自 GC 细胞系的 CRISPR 敲除数据,结合异常值分析,对细胞内靶点进行优先排序,以识别过表达或过度激活的依赖性靶点(图 7F),因为细胞内靶点通常需要肿瘤细胞依赖性才能发挥小分子作用。他们鉴定了21个候选靶点,包括 CDK6(一种已获批准的肿瘤药物的靶点)、12 个实验性药物的靶点以及 7 个具有小分子开发潜力的其他蛋白质,并将异常激活与功能脆弱性联系起来。
将优先靶点映射到生态型、基因组亚型和谱系背景,揭示了广泛的、特定背景下的治疗机会(图7G-H)。C1和C2富集的靶点(例如ASPN、MFAP4、OGN、COL14A1、DCN、PRELP、SERPINE2、COL3A1、BGN、COL5A1和LTBP1/2)主要在成纤维细胞中表达。相比之下,C4和C5富集的靶点则表现出更广泛的谱系分布。上皮细胞主要表达的靶点包括LAMC2和EPCAM(C4)、LAMA3(C5)、TSPAN8(MSI)和TROP2(CIN)。值得注意的是,在CIN和C4中富集的ERBB2在上皮细胞和成纤维细胞中均有mRNA表达(图7H)。这些结果共同表明,GC的治疗脆弱性取决于生态型和谱系,不仅限于上皮肿瘤细胞,还延伸到基质区室,并强调了制定以生态系统为依据的靶向策略的必要性。
图7. GC 异质性背景下的治疗靶点发现。
(A) ADC靶点、抗体靶点、T细胞靶点和其他非必需基因表面蛋白的箱线图。(B) 非必需表面蛋白的位点特异性糖型按高异常值频率排序。(C) 按聚糖类型划分的具有≥15个高异常值的位点特异性糖型比例。(D) 非必需表面蛋白按具有高位点特异性糖型异常值的样本数量排序。(E) 非必需表面蛋白按具有高位点特异性糖型异常值的样本数量排序。(F) 具有≥10个高异常值的假定细胞内靶点及其对应的CRISPR依赖性的箱线图。(G) 具有≥10个高异常值的假定细胞内靶点及其对应的CRISPR依赖性的箱线图。(H) 来自已发表的数据集的12种细胞类型中表达。
+ + + + + + + + + + +
结 论
本研究构建了一个包含15个组学层的多组学图谱,整合了来自159例原发性胃腺癌及其30例匹配的正常癌旁组织的基因组学、表观基因组学、转录组学、蛋白质组学、多种PTM、蛋白质-蛋白质相互作用、代谢组学和微生物组数据。利用细胞状态反卷积技术,定义了肿瘤生态型,通过捕捉与临床结果和免疫治疗反应相关的独特肿瘤微环境结构,细化了基因组和组织学亚型。多组学整合优先考虑基因组和表观基因组异常及其相关脆弱性;定义了生态型特异性的转录程序、信号通路、PTM、蛋白质相互作用网络和代谢调控;并鉴定了与生态型和耐药通路相关的微生物组特征。进一步利用肿瘤微环境背景下的蛋白质组学和PTM分析,优先筛选生态型、基因组亚型和细胞类型特异性的靶向蛋白。
+ + + + +



