

 English
English文献解读|Nat Commun(15.7):小鼠脱髓鞘模型和多发性硬化病变的比较转录组分析
✦ +
+
论文ID
原名:A comparative transcriptomic analysis of mouse demyelination models and multiple sclerosis lesions
译名:小鼠脱髓鞘模型和多发性硬化病变的比较转录组分析
期刊:Nature Communications
影响因子:15.7
发表时间:2026.05.18
DOI号:10.1038/s41467-026-72383-y
背 景
脱髓鞘,即神经元轴突周围髓鞘的丢失,是神经退行性疾病的主要驱动因素。在年轻健康的中枢神经系统(CNS)中,脱髓鞘可自发修复,足以提供神经保护并延缓疾病进展。然而,在多发性硬化症(MS)中,髓鞘修复的证据差异很大,且通常不完全,目前尚无获批的疗法可直接促进髓鞘再生。因此,了解其修复的细胞和分子机制迫在眉睫。毒素诱导的脱髓鞘模型广泛用于研究这些机制,因为它们能够提供时间可控且可重复的损伤。最常用的模型是溶血卵磷脂(LPC)注射和cuprizone (CPZ)饮食给药。LPC是一种去污剂,可造成局灶性损伤,该损伤会在大约4周内经历可重复的脱髓鞘、反应性胶质增生和少突胶质细胞前体细胞(OPC)分化为少突胶质细胞(OL)的阶段,从而实现修复。CPZ是一种铜螯合剂,给小鼠喂食4-6周后,可诱导大脑灰质和白质(GM和WM)中OL死亡。停用CPZ后,大脑会在2-3周内自发发生OPC分化和髓鞘再生。研究人员经常交替使用这两种毒性模型,选择特定方法更多是出于实验设计限制而非科学依据。然而,目前尚不清楚LPC和CPZ是否能诱导类似的分子变化和细胞状态。
实验设计
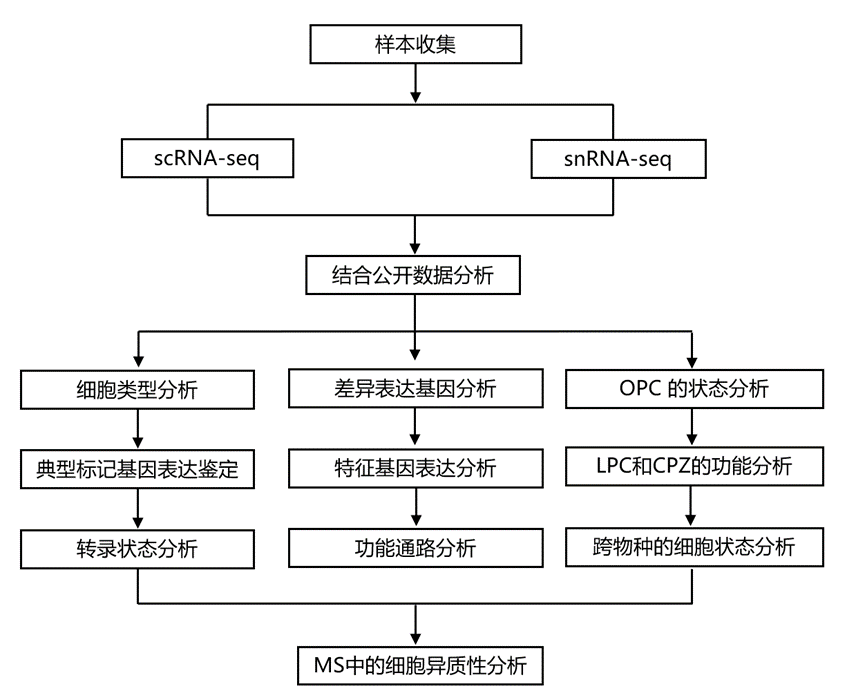
结 果
01

来自小鼠脱髓鞘模型和多发性硬化症患者样本的整合单细胞和单细胞核转录组数据集
为了比较LPC和CPZ诱导的脱髓鞘对不同白质细胞的影响,研究团队对成年小鼠胼胝体进行了snRNA测序,并将本研究数据与另外两项已发表的关于LPC或CPZ诱导脱髓鞘后胼胝体的单细胞转录组分析(scRNA-seq)研究整合。这些数据集涵盖了从基线(稳态)到脱髓鞘和随后的髓鞘再生的整个时间进程(图 1a)。值得注意的是,单细胞核转录组分析(snRNA-seq)样本中的LPC基线对照组注射了生理盐水,而scRNA-seq数据集中的对照组则未注射生理盐水。他们获得了112017个高质量的细胞和细胞核,完全整合的UMAP图谱显示,数据集和转录组类型之间存在高度重叠,同时保持了清晰的细胞类型分离(图 1b)。利用已知标记基因的表达,对对应于少突胶质细胞谱系细胞[OPC、定向少突胶质细胞前体细胞 (COP) 和成熟少突胶质细胞 (MOL)]、小胶质细胞、血管周围巨噬细胞 (PVM)、星形胶质细胞、神经祖细胞 (NPC)、室管膜细胞、内皮细胞、血管细胞、周细胞、T 细胞以及三个正在进行细胞分裂的细胞聚类(OPC、小胶质细胞和 NPC)进行了注释(图 1c)。MOL 和小胶质细胞构成捕获细胞的大多数(图 1c)。
按条件和时间点分层的UMAP可视化结果显示,LPC和CPZ模型中细胞类型丰度和转录状态发生了变化。正如预期,两种模型在脱髓鞘阶段均表现出MOL细胞显著减少和小胶质细胞增加(图 1d)。虽然已知星形胶质细胞在两种脱髓鞘模型中均有显著反应,但这些数据集中的星形胶质细胞数量不足以进行可靠的后续分析。值得注意的是,PVM细胞在LPC模型中特异性富集,表明LPC和CPZ诱导的反应在细胞水平上存在差异。使用scCODA对组成变化进行统计检验,他们发现LPC脱髓鞘的特征是PVM和小胶质细胞数量增加以及MOL细胞减少。在 LPC 髓鞘再生时间点,小胶质细胞数量持续升高,同时 T 细胞和 MOL 数量增加,而Dcx+ NPC 数量略有下降。在 CPZ 模型中,脱髓鞘和髓鞘再生过程中小胶质细胞数量也显著增加,而 MOL 的减少仅限于脱髓鞘阶段。
为了构建可比的人类队列,他们还重新分析了近期发表的snRNA-seq数据集,仅从其图谱中提取了白质样本。总共,人类数据包括来自非MS患者的13个对照组白质(WM)样本、17个外观正常的白质(NAWM)样本、21个活动性病灶(AL)、17个慢性活动性病灶(CAL)、13个慢性非活动性病灶(CIL)和11个髓鞘再生病灶(RL),共涉及47名个体(图 1e)。年龄、性别(25名女性,22名男性)、死后间隔(PMI)或每个细胞核的计数均无显著差异。从许多个体中收集了多个病灶,共计 92 个样本,其中大部分病灶样本来自患有进展型疾病的患者——继发性进展型多发性硬化症 (SPMS) 和原发性进展型多发性硬化症 (PPMS)。
经过质量过滤后,他们整合了321565个细胞核,并使用经典标记对人类细胞类型进行了注释,重现了原始研究中报道的主要细胞类别(图 1f-g)。与小鼠数据类似,人类白质图谱主要由神经胶质细胞组成,特别是MOL、OL和星形胶质细胞。按病变类型分层的UMAP分析显示,小胶质细胞、星形胶质细胞和MOL的转录状态发生了变化(图 1h),scCODA分析显示,在AL和CAL中,MOL的丰度显著降低。外周免疫细胞,包括T细胞和B细胞,主要在MS病变样本中检测到,而在WM中则基本缺失,这与它们在MS中已知的病理作用相符。这些整合的小鼠脱髓鞘和人类 MS 数据集共同重现了预期的细胞和组成变化,并为脱髓鞘和髓鞘再生的比较转录组分析提供了一个协调的跨物种框架。
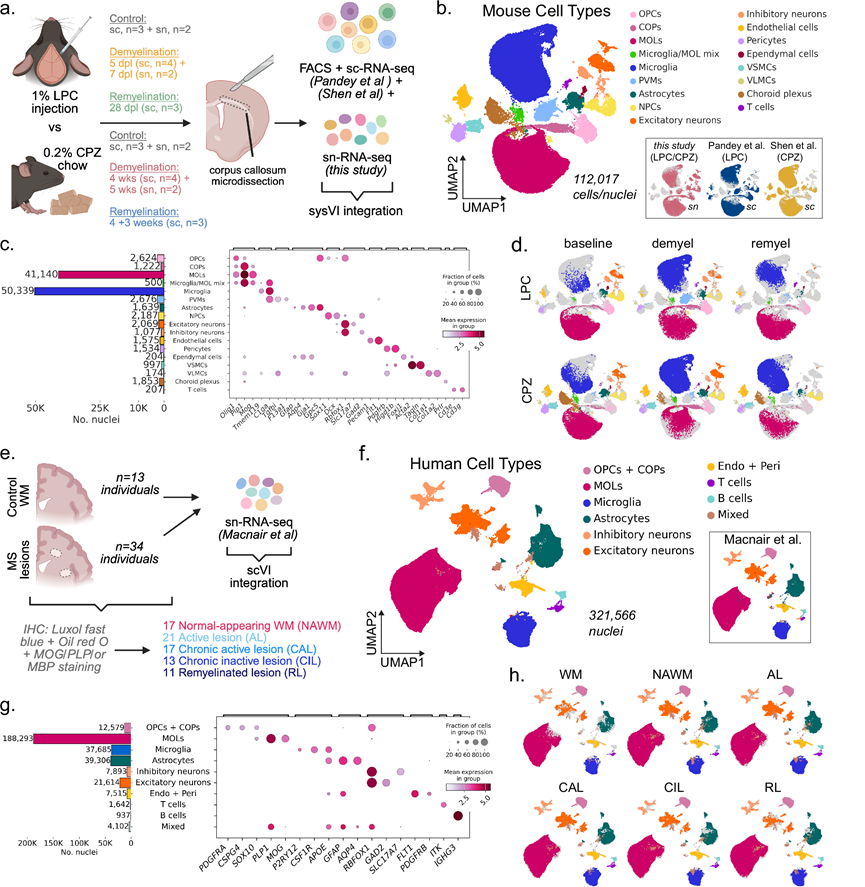
图1. 来自小鼠脱髓鞘模型和 MS 组织的单细胞和snRNA-seq 数据集。
(a) 本研究中包含的小鼠数据集示意图。(b) 整合的小鼠数据集的UMAP嵌入图,按细胞类型注释。(c) 条形图(左)显示了细胞类型组成,散点图(右)显示了典型标记基因的表达。(d) UMAP显示了不同处理(LPC或CPZ)和实验阶段的细胞类型分布。(e) 人类snRNA-seq数据集概述。(f) 整合的人类数据集的 UMAP 嵌入,并按主要细胞类型进行注释。(g) 条形图(左)显示了细胞类型组成,散点图(右)显示了典型标记基因表达。(h) 根据每种病变类型的细胞类型注释着色的 UMAP。
02
CPZ诱导了一种独特的DAO状态,这种状态在LPC脱髓鞘中并不存在
为了明确OL谱系细胞对LPC和CPZ脱髓鞘的反应,他们对整合的小鼠sn/scRNA-seq数据集中的OL谱系细胞进行了亚聚类分析(图 1b),并使用已知的OPC、少突胶质细胞前体细胞(COP)、新生少突胶质细胞(NFOL)、髓鞘形成少突胶质细胞(MFOL)和髓鞘形成MOL标记物对其进行了重新注释(图 2a-b)。PAGA连接性重现了祖细胞向成熟状态的已知发育进程,推断出从MFOL到MOL_A的连接性。他们定义了四种稳态小鼠MOL状态(MOL_A-D),它们都表达成熟标记物(Plp1、Mog、Mag、Opalin),以及聚类特异性标记物(MOL_A:Il33低;MOL_B:Il33高和Ptgds高;MOL_C:Selenop、Spock3、Anln表达增加;以及MOL_D:Klk6、S100b、Anxa5),这些标记物在之前的研究中已有分析数据(图 2b)。MOL_B和MOL_C构成成年小鼠胼胝体少突胶质细胞的大多数,在基线时占MOL的75-80%,这与之前关于成年小鼠胼胝体中Klk6低表达的报道一致。此外,他们还鉴定了三个 MOL 亚聚类,它们表达与先前描述的 DAO 状态相关的基因,包括Serpina3n、C4b和H2-D1(MOL_E和 F)以及Cdkn1a、Ddit3和Nupr1(MOL_G)(图 2b)。
他们假设在脱髓鞘过程中,疾病相关少突胶质细胞(DAO)状态会以模型特异性的方式选择性富集。为了定量差异丰度(DA),他们应用了Milo模型,该模型在k近邻图上模拟部分重叠的局部细胞邻域(nhoods)。LPC和CPZ均在脱髓鞘过程中诱导了广泛的MOL耗竭和祖细胞的适度增加(图 2c)。为了直接比较CPZ和LPC的细胞组成,他们进行了单独的统计检验,比较了nhoods的分布。按细胞类型和条件汇总了上调、下调和非显著性nhoods的计数,并分别针对脱髓鞘和髓鞘再生时间点进行了统计检验。LPC诱导的脱髓鞘优先富集DAO的MOL_E和F状态,而CPZ诱导的脱髓鞘则导致DAO的MOL_G状态积累(图 2d)。有趣的是,髓鞘再生后的LPC样本显示稳态MOL细胞群几乎完全恢复,而CPZ样本则表现出持续的MOL耗竭,并且两种髓鞘再生条件下MOL_E DAO细胞均持续增加。
为了确定LPC和CPZ诱导的脱髓鞘MOL状态的转录程序,他们进行了伪批量差异表达分析,比较了脱髓鞘和髓鞘再生状态下所有MOL与其各自基线水平的差异表达。CPZ诱导的髓鞘再生样本表现出较高的样本间异质性,因此排除在本分析之外。LPC在脱髓鞘和髓鞘再生时间点诱导的差异表达基因(DEG)数量相似(分别为902个和1044个基因),而CPZ诱导的脱髓鞘则诱导了更为显著的转录变化(4230个DEGs),表明MOL身份发生了更深层次的重编程。利用基因集富集分析(GSEA)对所有DEG分析中检测的基因进行排序,结果显示,TNFα通过NFκB和干扰素(IFN)通路发出信号是不同条件下共同的反应,且Gadd45a/b、Stat1/3、B2m、H2-D1和Socs3基因均上调(图 2e-g)。LPC诱导的MOL差异表达基因尤其富集于IFNγ和IFNα信号通路,表现为Irf1、Ifi27、Ifi35和Stat2基因上调。相比之下,CPZ 脱髓鞘特异性地富集了 Myc 靶基因(Eif4e、Eif4a1、Odc1)、DNA 损伤和修复基因(Tp53、Ddb1、Pcna)以及未折叠蛋白反应 (UPR) 特征基因(Atf4、Hspa5)(图 2e-g)。这些特征表明蛋白质合成增加以及基因毒性和内质网应激反应失调,这与先前在 CPZ 处理的少突胶质细胞中描述的氧化损伤和代谢毒性一致。
为了验证CPZ特异性DAO特征在原位的表达,他们对两个高表达的MOL_G基因Cdkn1a和Nupr1进行了RNAscope分析。Cdkn1a编码p21,一种p53依赖性激酶抑制剂,在DNA损伤后诱导表达;Nupr1编码一种参与氧化应激反应、DNA损伤和铁死亡的转录调节因子。在CPZ脱髓鞘5周后(5周),OL细胞中这两个转录本均显著上调,并在3周恢复期(5+3周)后恢复至基线水平(图 2h-k)。相反,与注射生理盐水的对照组相比,在LPC注射后损伤后7天(dpl)或28天(分别为脱髓鞘和髓鞘再生期),OL细胞中Cdkn1a和Nupr1的表达均未发生显著改变(图 2h-k)。鉴于CPZ可导致持续数周的OL丢失,他们研究了LPC诱导的损伤中是否会发生早期应激反应。然而,对注射LPC后2天(2 dpl)的脑组织进行分析,未检测到Cdkn1a或Nupr1的表达(图 2l-m)。综上所述,这些数据表明CPZ脱髓鞘诱导了一种独特的、与应激相关的DAO状态,其特征是DNA损伤和UPR激活,而这种状态在炎症性LPC模型中并未观察到。
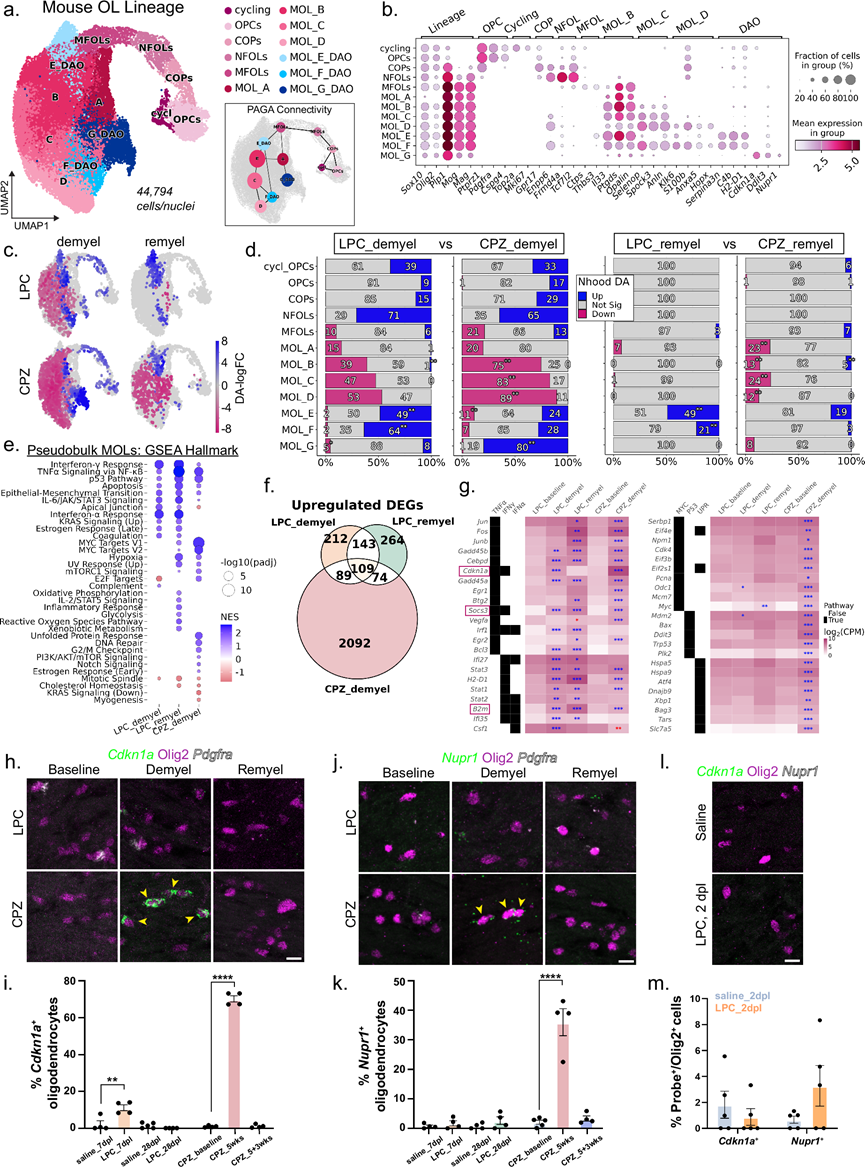
图2. CPZ 诱导少突胶质细胞产生独特的转录状态。
(a) 小鼠少突胶质细胞谱系细胞重聚后的 UMAP 嵌入图。(b) 谱系和聚类特异性标记基因的散点图。(c) 差异表达细胞邻域叠加在 UMAP 嵌入图上。(d) 条形图显示了按细胞类型注释的增加、减少或不变的邻域的比例。(e) 散点图显示了从伪批量 MOL 的差异表达测试中获得的 Hallmark 通路富集情况。(f) Venn 图展示了共有和条件特异性的显著上调基因。(g) 分别显示细胞因子相关基因表达(左)和MYC V1、p53通路及UPR相关基因表达(右)。(h, j, l) 分别为小鼠胼胝体中Cdkn1a、Nupr1或Cdkn1a和Nupr1均 表达的代表性RNAscope图像。(i, k, m) 细胞的百分比
03
在髓鞘再生阶段,LPC 和 CPZ 趋同于一种共同的 DAO 状态,该状态上调免疫相关基因的表达
他们对脱髓鞘和修复过程中MOL状态动态的分析表明,在LPC和CPZ模型中,MOL_E DAO聚类均保持高表达。为了验证这种共同的DAO状态的存在,他们对MOL_E中高度富集的两个基因Socs3和B2m进行了RNAscope分析。Socs3编码细胞因子信号抑制因子(SOCS),该因子负调控免疫反应和炎症;B2m编码I类主要组织相容性复合体(MHC-I)的不变轻链。与各自的对照组相比,注射LPC和喂食CPZ的小鼠OL中 Socs3和B2m均显著上调(图3a-d)。在LPC模型中,从脱髓鞘到髓鞘再生,Socs3 +和B2m+ OL的比例保持相似,而在CPZ模型中,这两个比例从脱髓鞘到髓鞘再生逐渐增加(图 3b-d)。虽然B2m+少突胶质细胞在 5 周和 5+3 周之间没有显著差异,但这些趋势表明免疫相关的少突胶质细胞程序在恢复过程中逐渐出现。他们还研究了 DAO 的空间分布,尤其是在诱导弥漫性脱髓鞘的 CPZ 模型中。Cdkn1a+ MOL在 CPZ 暴露 5 周时显著诱导,并在 5+3 周时消退,主要定位于胼胝体,在深部灰质和纹状体中含量极少。相比之下,Socs3+ DAO 在脱髓鞘时稀少,但在髓鞘再生过程中增加,广泛分布于整个白质,与广泛的Mbp丢失和 Iba1+小胶质细胞聚集平行。在 CPZ 病变中未检测到 DAO 丰度的显著内侧-外侧差异。在局灶性 LPC 模型中,Socs3+ DAO局限于病变核心,在 7 dpl 时出现频率较低,到 28 dpl 时出现频率增加。
由于在稳态条件下DAO细胞群不存在,他们推测它们要么来源于受损的OL,要么来源于新生成的、采用类似DAO转录程序的OL。支持后一种可能性的是,在PAGA分析中,MFOL与MOL_E显示出直接连接(图 2a),这表明脱髓鞘改变了OL的成熟轨迹,而不是诱导不可逆的损伤。为了确定免疫相关的DAO标记物是否在髓鞘再生过程中由新生成的OL表达,他们在脱髓鞘过程中进行了EdU标记,并分析了每个模型在两个髓鞘再生阶段的脑组织(图 3e)。大多数EdU标记的细胞表达OL谱系标记物Olig2,而OPC标记物Pdgfra的表达极少,证实了新生成的OL的成功标记(图 3f-h)。值得注意的是,在两种模型中,大多数 Olig2+ EdU+细胞均表达Socs3(图 3i)。有趣的是,在 LPC 注射后 42 天(即脱髓鞘时间点后 5 周),新生成的少突胶质细胞中Socs3 的表达持续存在,但在停用 CPZ 5 周后显著下降(图 3i),这表明两种模型中免疫相关少OL状态的时间分辨率存在细微差异。
在LPC和CPZ模型中,MOL均上调保守的干扰素和应激相关基因特征,包括MHC-I抗原呈递基因(H2-D1、H2-K1、B2m、Tap2、Nlrc5)、细胞因子信号传导介质(Stat1/2/3、Irf1/3/9)和干扰素刺激基因(Ifi27、Rtp4、Zc3hav1)。这种状态伴随着补体和胶质细胞应激标志物(Serpina3n、C4b、Gadd45a/b、Socs3)以及细胞骨架重塑因子(Vim、Anxa2、Cd63)的上调,表明MOL对炎症信号存在积极的适应性反应,类似于实验性自身免疫性脑脊髓炎(EAE)中观察到的情况。值得注意的是,既往研究已描述了在EAE小鼠模型和MS死后脑组织中OL谱系细胞内免疫相关基因的上调。为了直接比较这些反应,他们分析了LPC和CPZ诱导的脱髓鞘中DEG的表达情况,并将其与先前发表的基于EAE小鼠脊髓分离的OL谱系细胞scRNA-seq生成的DEG列表进行比较。EAE中上调的DEG分别有27%和22%与LPC和CPZ诱导的脱髓鞘中OL谱系细胞的表达重叠。相比之下,CPZ和EAE中下调的DEG重叠数量(23%)显著高于LPC组(7.5%)。EAE 和CPZ 诱导的少突胶质细胞 (MOL) 均下调了代谢和膜维持基因(Slc1a3、Abhd12、Gprc5b、Stard5)以及结构或黏附因子(Ncam1、Dclk1、Tspan15)的表达,表明 CPZ 和 EAE 诱导的成熟少突胶质细胞表型不稳定,代谢韧性降低。有趣的是,EAE 和 CPZ 诱导的 MOL 还下调了表观遗传修饰因子(Dnmt3a、Hdac11、Tet1)的表达,这可能导致 EAE 和 CPZ 诱导的转录重塑范围较 LPC 诱导的更为广泛。
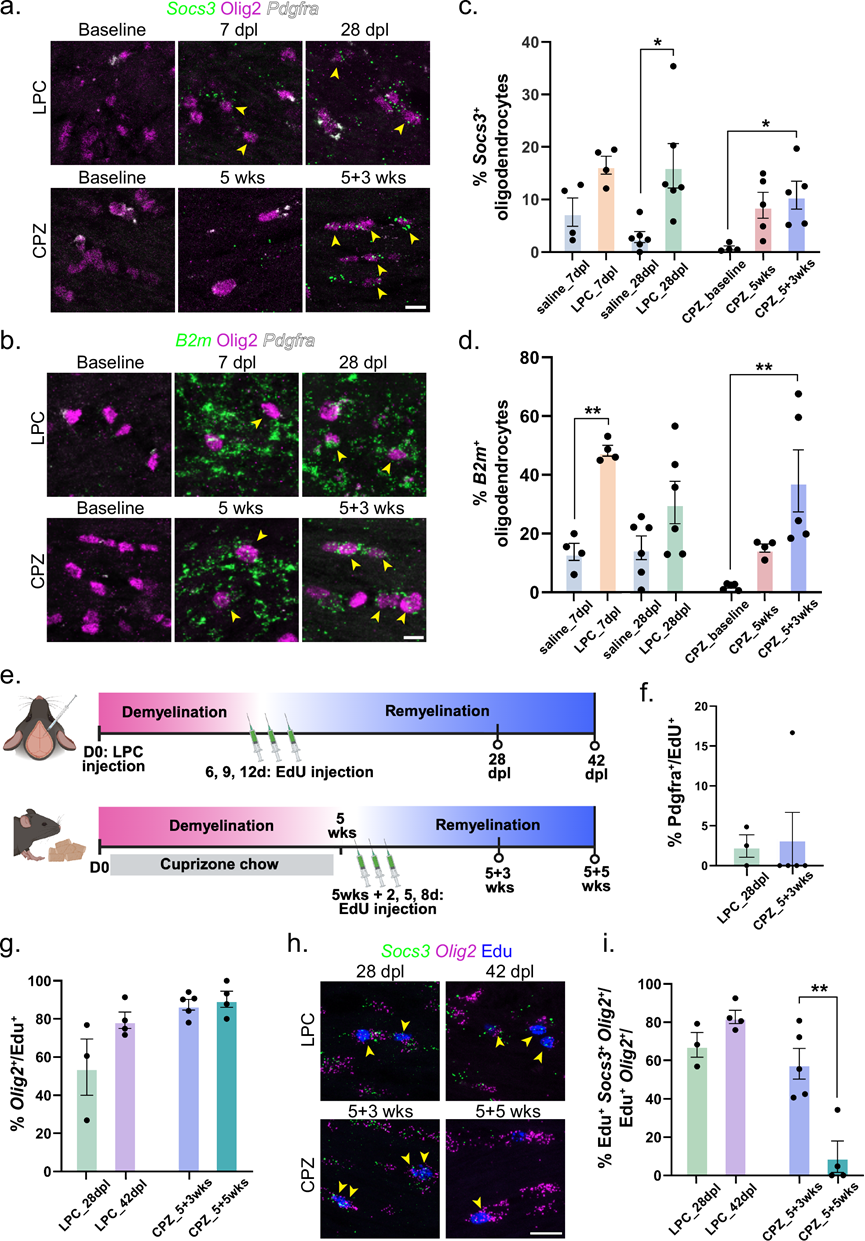
图3. 在 CPZ 和 LPC 模型中,髓鞘再生过程中均出现了表达免疫相关基因的趋同少突胶质细胞状态。
(a-b) 小鼠胼胝体的代表性RNAscope图像显示了Socs3或B2m的表达。(c-d)细胞百分比。(e) 示意图显示了 EdU 标记实验的时间线。(f) EdU+细胞(少突胶质细胞前体细胞,OPC)的百分比。(g) 表达Olig2的EdU+细胞的百分比。(h) 小鼠胼胝体的代表性RNAscope图像显示EdU+ Olig2+细胞中Socs3的表达。(i) 显示了注射LPC并喂食CPZ的小鼠胼胝体中表达Socs3的新生OL的百分比。
04
与LPC相比,CPZ脱髓鞘能更忠实地重现MS病变中观察到的DAO状态
在小鼠模型中鉴定出不同的DAO状态后,他们研究了MS病变中是否存在类似的状态,对图 1中snRNA-seq MS WM数据集的人类OL谱系细胞进行了亚聚类和重新注释(图 4a)。利用已建立的标记表达,鉴定了人类OPC、COP和4种MOL细胞群(MOL_A–D)(图 4b)。人类MOL状态由聚类特异性标记物定义(MOL_A:RBFOX1和SLC5A11高 表达;MOL_B:表达MOL_A标记物,CTNND2和PLXDC2低表达;MOL_C:RBFOX1和SLC5A11表达缺失,CTNND2和PLXDC2高表达,OPALIN和LAMA2表达上调; MOL_D:与MOL_C相似,但FTL和CSPRP1表达上调)。此外,还鉴定了表达经典DAO相关标记物的聚类(人类MOL_E-G)。具体而言,人类MOL_E高表达HSPH1、FOS和JUN,人类MOL_F以CDKN1A、NEDD4L和GADD45B表达为特征,而人类MOL_G低表达GPC5和CD63。使用来自先前 MS 研究的基因特征验证了DAO 注释,结果显示人类 MOL_E–F 和 OPC 聚类中 DAO 基因的富集情况一致。PAGA连接性分析揭示了从 OPC 到 COP 的主要分化轨迹,以及从 MOL_A 到 B、C 和 D 的顺序进展,此外还有连接 DAO 状态的额外连接,这与应激诱导的偏离稳态谱系路径一致。
为了定量MS不同病症下OL谱系组成的变化,他们使用Milo软件进行分析。与对照组白质(WM)相比,正常外观白质(NAWM)中稳态MOL_A-D细胞群基本保持不变。相反,MS病变中稳态MOL状态表现出不同程度但显著的改变,其中RLs的变化最少(图 4c)。所有MS病变类型均显著富集人类DAO状态MOL_F和MOL_G,但MOL_F的丰度明显低于MOL_G(MOL_F平均每个样本33个细胞,而MOL_G平均每个样本245个细胞)(图 4c)。有趣的是,相对较小的MOL_E细胞聚类在两个对照组白质样本中高度富集(占细胞总数的44%),这两个样本均来自85岁高龄的个体。人类 MOL_E 与 MS 富集的 DAO 共有基因表达变化,表明生理衰老可能会在 MOL 中诱发类似于脱髓鞘相关转录变化的应激状态。
为了评估MS不同病变类型之间的转录组改变,进行了DEG分析,比较了每种MS病变类型的MOL与WM。CAL的转录组扰动最大(1629个基因),而NAWM的变化最少(328个基因),且不同病变类型之间存在显著的重叠。通路富集分析显示,在多种MS病变类型中,TNFα信号通路(NFKB1A、BIRC3、CEBPD)、p53通路(IER5、CDKN2A、CDKN2B)和IFNα反应(IRF7)均持续上调(图 4d)。这些炎症和应激反应通路与小鼠脱髓鞘模型中观察到的通路相似。CIL 和 RL 病变共有其他通路,包括 TGFβ 和 PI3K/AKT/mTOR 信号通路,表明病变阶段特异性重塑程序的获得。
为了直接比较小鼠和人类OL谱系细胞,他们应用了MetaNeighbor分析。细胞类型聚类为两个主要的跨物种组,分别对应于祖细胞和中髓鞘MOL。在MOL分支中,DAO形成了一个独特的亚聚类,与稳态MOL分离,表明其转录特征在不同物种间保守。为了更好地了解哪些基因驱动了DAO之间的相似性,接下来定量了小鼠毒性模型和人类MS病变之间共有的DEG。LPC脱髓鞘MOL与MS MOL共有22-47个上调的DEG,而CPZ脱髓鞘MOL共有25-60个(图 4e)。具体而言,在CIL和/或RL病变中,多种小鼠DAO基因表达上调,包括SOCS3、CDKN1A、FOS、JUNB和GADD45A/B。NUPR1在CAL中也表达上调,但未达到他们的显著性阈值。有趣的是,B2M在任何MS病变类型中均未因MOL而显著上调,然而在其他MS数据集中,B2M已证实表达上调。所有三个数据集的交集鉴定出37个基因,这些基因无论脱髓鞘机制如何均显著上调,且富集于细胞骨架重塑和应激反应程序。共有41个基因独特地与至少一种MS病变类型和脱髓鞘时的LPC MOL重叠,其中包括参与神经保护(GDNF)、脂质代谢和胆固醇转运(APOE)、谷氨酸能和轴突-胶质细胞Ephrin信号通路(GRM8、EPHA4)、干扰素信号通路和MHC-I调控(IRF1、NLRC5)以及细胞外基质和细胞黏附分子(TNR、CNTN3、ITGA8)的基因。CPZ特异性重叠基因包括106个基因,主要由应激反应转录因子(KLF15、KLF9、KLF10、MAFK、CEBPG、JUNB和FOS)组成。CPZ基因集包含多个参与少突胶质细胞命运决定和分化阻滞的基因(ID4、NGFR、EGR1、SESN2、RUNX2)以及抗凋亡因子BCL2。当检测重叠下调基因的数量时,发现CPZ脱髓鞘中的MOL与MS病变的重叠基因数量(137个)远高于LPC(7个),并且在所有3种状态下仅有12个基因是共同的(图 4e)。CPZ特异性重叠下调基因包括OL稳态和髓鞘完整性的关键介质,例如MOG。MAG、OMG、PLLP和GJB1。其他 MS/CPZ 特异性下调基因进一步支持OL功能的广泛受损,包括神经调节蛋白共受体ERBB3(OL 存活和分化)、瓜氨酸化酶PADI2(髓鞘碱性蛋白的翻译后修饰)、脂质生物合成基因ST3GAL5和LPCAT2、促存活受体LPAR1以及分化相关转录因子E2F3。髓鞘结构和代谢基因的下调表明,CPZ 诱导和 MS 相关的 DAO 在维持髓鞘稳定方面均存在功能障碍。
最后,为了检验小鼠DAO的不同状态是否与人类DAO相对应,他们构建了代表CPZ特异性DAO状态(小鼠MOL_G)和髓鞘再生DAO状态(小鼠MOL_E)的基因特征。CPZ特征在人类DAO聚类中显著富集,支持了CPZ脱髓鞘模型与损伤相关DAO转录状态高度相似的结论(图 4f)。CPZ相关基因特征包括应激反应基因(ATF4、DDIT3、NUPR1)、缺氧和损伤反应基因(VEGFA、SERPINE1)以及促炎介质(CD44、LGALS3)。小鼠髓鞘再生特征在人类DAO聚类中也有富集,但是程度较轻,这些特征包括免疫信号负调控因子(SOCS3、BCL3、TNFRSF1A、GADD45B/G)和膜/细胞外基质重塑基因(CD63、ANXA2、SNX10)。基因评分的热图显示,CPZ特征在MOL_G中富集,髓鞘再生特征在MOL_E中富集,并且人类DAO也富集,尤其是在MOL_F中。总的来说,这些分析表明,两种小鼠脱髓鞘模型均能重现MS病变中DAO的关键特征,包括TNFα和干扰素信号通路的增强。然而,CPZ脱髓鞘模型更忠实地重现了人类DAO的转录程序,特别是髓鞘和稳态基因的下调。重要的是,这些发现确立了SOCS3和CDKN1A是跨物种 DAO 状态的保守元件,值得进一步从机制上研究它们在 OL 病理中的作用。
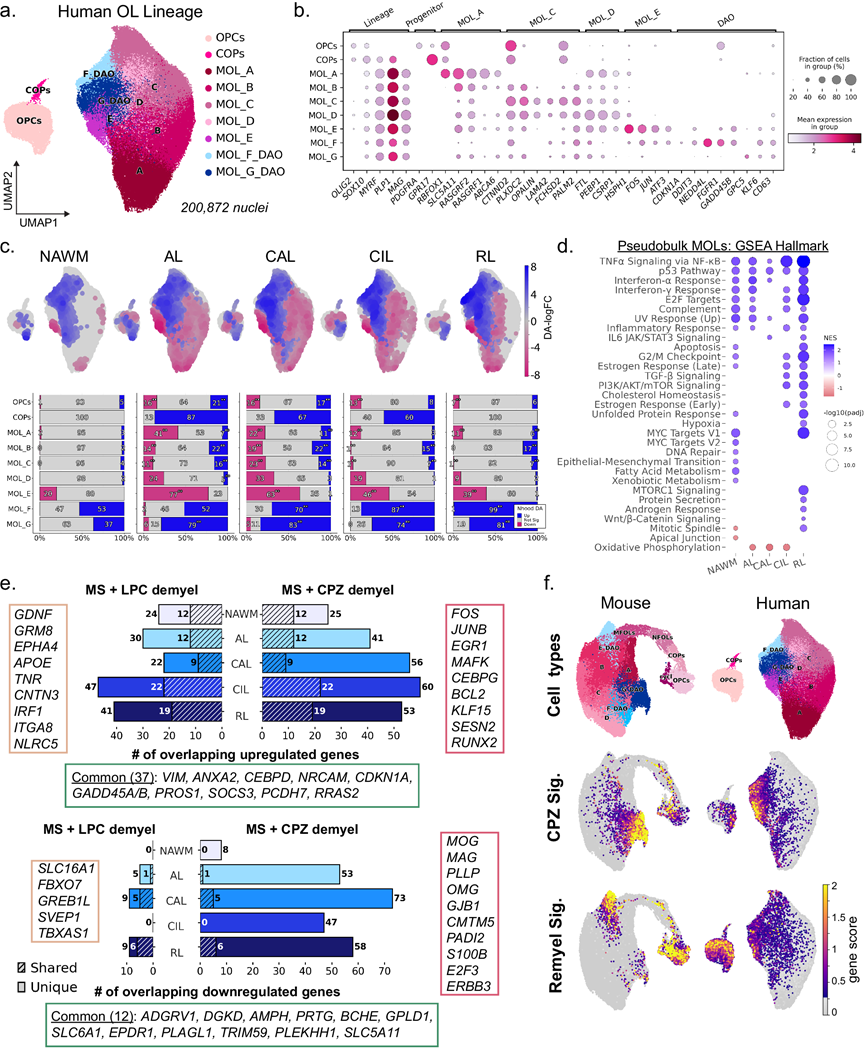
图4. 小鼠脱髓鞘模型与人类 MS 病变中的疾病相关少突胶质细胞具有相似的转录表型。
(a) 重新聚类的人类少突胶质细胞谱系细胞的 UMAP 嵌入图。(b) 谱系和聚类特异性标记基因的散点图。(c) 差异表达细胞邻域叠加在 UMAP 嵌入图上(上图)。柱状图(下图)显示了与 NAWM 相比,按细胞类型注释的增加、减少或不变的 nhoods 的比例。(d) 散点图显示了基于每种病变类型的伪块状 MOL 的差异表达测试计算的 Hallmark 通路富集情况。(e) 条形图显示了脱髓鞘时 LPC 和 CPZ 中上调和下调的差异表达基因 (DEG) 与 MS 病变类型的重叠情况。(f) UMAP 图显示了小鼠和人类少突胶质细胞(OL) 谱系细胞,以及从 CPZ 相关和髓鞘再生相关的小鼠差异表达基因中获得的基因评分。
05
LPC 和CPZ脱髓鞘诱导的 DAM 状态在转录上相似,并持续到髓鞘再生期
除了OL谱系改变外,脱髓鞘还引起小胶质细胞数量和状态的显著变化(图 1d)。因此,他们探究了LPC和CPZ诱导的脱髓鞘是否会引发不同的小胶质细胞反应。对小鼠小胶质细胞和巨噬细胞进行重新聚类分析,鉴定出小胶质细胞亚群(Mg_A–E)、PVM以及增殖细胞群(图 5a)。利用先前定义的固有免疫基因集,分析了疾病相关小胶质细胞/巨噬细胞 (DAM)评分,该评分在高度相关的Mg_C–E亚群、PVM和增殖细胞中富集(图 5a)。这些亚群表达典型的DAM标记物(Apoe、Cd63、Lyz2),并下调稳态小胶质细胞基因(Tmem119和P2ry12)(图 5b)。PVM 的特征是表达谱系标记物C1qa和Trem2,并且缺乏小胶质细胞特异性基因,如Tmem119和Cx3cr1。值得注意的是,PVM 表达 DAM 标记物,并且富含免疫信号基因Igqap1、Ms4a7和Msrb1。
与CPZ脱髓鞘相比,LPC脱髓鞘中稳态小胶质细胞(Mg_A和Mg_B)的比例显著降低(图 5c-d),表明LPC注射后小胶质细胞活化更强。LPC和CPZ脱髓鞘均使DAM(Mg_C-E)的比例增加至相似水平,但LPC诱导的循环小胶质细胞比例显著更高(图 5d)。此外,PVM仅在LPC病变中富集,他们通过定量分析健康和脱髓鞘白质中Cd206高表达的血管周围细胞验证了这一点(图 5e)。循环小胶质细胞和PVM仅限于脱髓鞘时间点,并在髓鞘再生过程中消失。相反,在两种模型中,DAM聚类在髓鞘再生过程中均持续存在,但与CPZ组相比,LPC组中的DAM聚类显著升高(图 5d)。为了确定DAM是否长期存在,他们对脱髓鞘和髓鞘再生晚期(LPC注射后42天,CPZ停药后5周) 表达ApoE的Cd11b +小胶质细胞进行了定量分析。LPC组后ApoE⁺小胶质细胞仍然丰富,而CPZ组后其表达恢复至基线水平,表明LPC模型中小胶质细胞活化的消退延迟(图5f)。重要的是,他们通过将每个数据集与其各自的队列进行整合,证实了LPC和CPZ模型中DAO和DAM状态的可重复富集,且与数据集来源或测序平台无关。
接下来,他们使用伪批量分析比较了小胶质细胞的基因表达变化,将脱髓鞘和髓鞘再生的小胶质细胞与其各自的基线对照组进行比较。由于基线样本中不存在 PVM,因此将其与 LPC 稳态小胶质细胞进行比较。所有比较均显示出广泛的转录重塑,其中 LPC 脱髓鞘小胶质细胞和 PVM 中DEG的数量最多。基因集富集分析发现 LPC 和 CPZ 诱导的 DAM/PVM 之间存在多个共同激活的通路,包括胆固醇稳态、上皮-间质转化和活性氧信号通路(图 5g)。LPC 髓鞘再生和 CPZ 脱髓鞘 DAM 还富集了免疫反应通路,包括 IFNα 和 IL-6 信号通路(图 5g)。比较结果显示,在两种模型和时间点上,DAM中共有136个基因持续上调,其中包括一些已知的DAM标志物,例如Apoe、Lgals3和Spp1(图 5h)。CPZ DAM中约67%的上调基因也在LPC DAM中诱导表达,而LPC上调基因中仅有22-27%与CPZ重叠,这表明LPC特异性炎症反应更为广泛。LPC特异性基因包括一些免疫相关介质,例如Gpnmb、Ms4a7和Mmp12。下调基因的保守性更高,在所有条件下均有941个基因表达降低,包括核心稳态小胶质细胞基因(P2ry12、Cx3cr1)和吞噬抑制因子(Cd33、Gpr34),表明不同模型中稳态特性的丧失高度一致(图 5h,补充图 13c)。有趣的是,LPC髓鞘再生DAMs仍然富集炎症基因(Il1b、Cxcl9、Cd69)、补体成分(C1qa、C3)和缺氧/糖酵解相关基因(Mt1、Slc2a1),提示LPC髓鞘再生阶段小胶质细胞持续处于促炎状态(图 5i)。这些研究结果表明,虽然两种模型都产生了保守的 DAM 程序,但 LPC 病变在髓鞘再生过程中维持了代谢活跃和促炎的小胶质细胞状态。
为了确定小胶质细胞对毒性脱髓鞘的反应是否与EAE中观察到的反应相似,他们将分析中的基因表达变化与先前发表的从EAE高峰期小鼠脑中分离的小胶质细胞的scRNA-seq数据集进行了比较。他们将数据子集化,仅包含未敲除CD83的细胞,并进行了伪批量基因表达分析,比较了EAE组和对照组的小胶质细胞。EAE差异表达结果的通路分析显示,干扰素信号通路、Myc靶基因和代谢转变均发生强烈诱导。在EAE、LPC和CPZ条件下,小胶质细胞均上调了一种保守的炎症和代谢激活程序,其特征是诱导细胞因子和趋化因子介质(IL-1β、CXCL10)、MHC-I相关抗原加工组分(IFI-30、PSMB-8、PSME-1/2、H2-T22)、氧化应激调节因子(CYB、TXN-1)以及糖酵解酶(LDHA、PKM)的表达。这种共同的反应伴随着稳态受体和代谢调节因子(包括P2RY13、GPR34、ADRB2和LPIN1)的下调。相比之下,LPC和CPZ诱导的转录重塑范围更广、程度更高,远超EAE脑小胶质细胞。毒素引起的脱髓鞘显著增强了脂质处理和胆固醇代谢程序(Apoe、Lpl、Abca1、Npc2)、吞噬溶酶体和蛋白水解机制(Gpnmb、Cd68、Ctsb、Ctsd)以及先天免疫信号网络(Tyrobp、Axl、Cd14、Tlr2),同时进一步抑制了经典的小胶质细胞特征基因(P2ry12、Tmem119、Cx3cr1、Sall1)。因此,虽然LPC和CPZ能够清晰地重现EAE中存在的核心激活程序,但它们驱动了一种数量上更强、性质上更广的状态,其特征是脂质加工增强、吞噬作用增强和代谢重编程。这种增强的转录反应与毒素模型中明显的脱髓鞘现象相一致,并强调了在所研究的时间点,EAE中的脑小胶质细胞表现出相对抑制的激活模式。
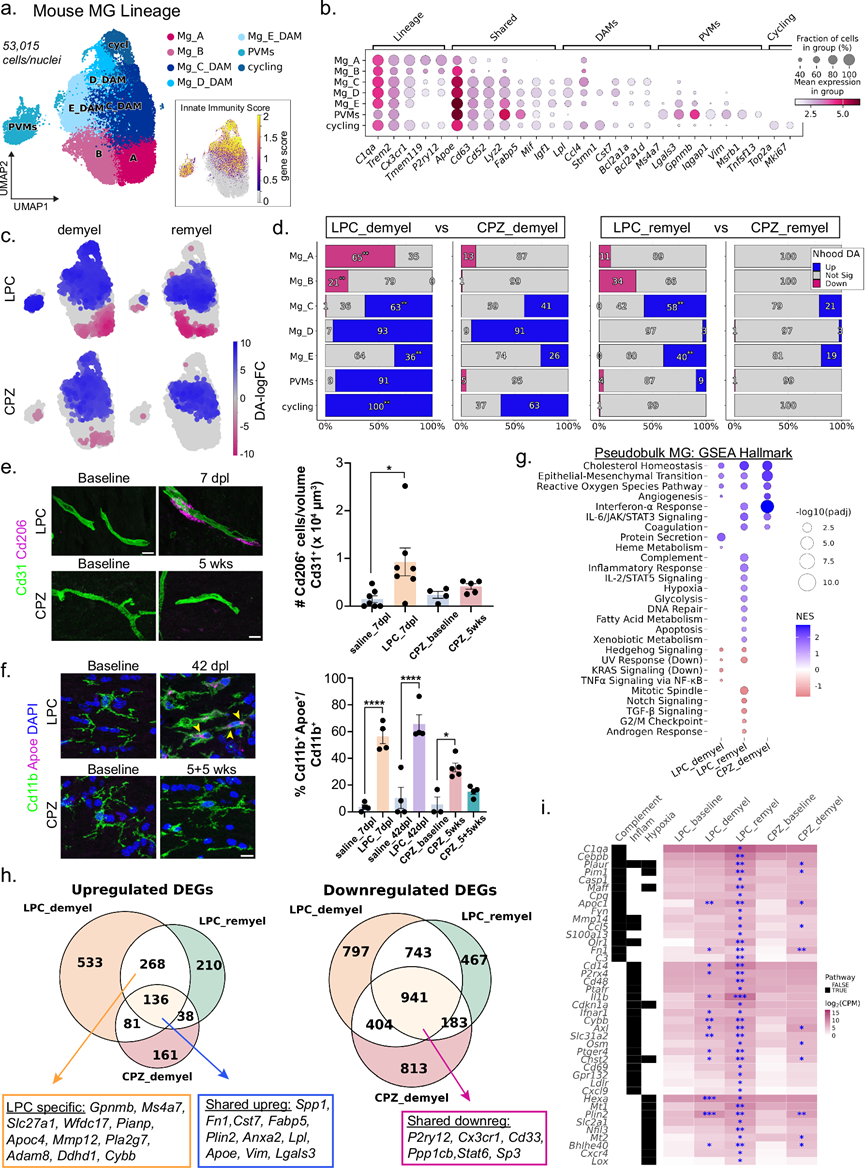
图5. 小鼠脱髓鞘模型中小胶质细胞的基因表达变化具有保守性,但 LPC 会导致炎症加剧。
(a) 小鼠巨噬细胞/小胶质细胞谱系细胞重聚后的 UMAP 嵌入图。(b) 不同细胞类型中谱系和聚类特异性标记基因的散点图。(c) 差异表达细胞邻域叠加在 UMAP 嵌入图。(d) 条形图显示了按细胞类型注释的增加、减少或不变的邻域的比例。(e) 代表性的 IHC 图像(左)显示了小鼠胼胝体中 Cd31 和 Cd206 的表达。(f) 左侧为代表性的免疫组化 (IHC) 图像,显示了小鼠胼胝体中 Cd11b 和 Apoe 的表达。右侧柱状图显示了Apoe+ 小胶质细胞百分比。(g) 点图显示了LPC或CPZ中假性大胶质细胞差异表达检测得到的Hallmark通路富集情况。(h) Venn图展示了在指定时间点上显著上调和下调的DEG的独特和重叠部分。(i) 热图显示了LPC髓鞘再生富集通路。
06
在MS中富集的小胶质细胞在不同类型的病变中表现出异质性状态
为了比较毒素诱导的小胶质细胞反应与MS病理中的人类小胶质细胞,他们对人类小胶质细胞进行了重新聚类,并鉴定出七个亚聚类(Mg_A–F 和 PVM)(图 6a)。促炎基因集的评分显示,Mg_A 中未见富集,Mg_E 中略有富集,而 Mg_F 中则显著富集,这表明 MS 中不同亚型的小胶质细胞激活状态存在异质性。人类 Mg_A 表达高水平的谱系定义基因和低水平的炎症标志物,归类为稳态小胶质细胞(图 6b)。相比之下,人类 Mg_B 和 Mg_C 保留了稳态标志物P2RY12的表达,并显示出 DAM 相关基因的轻微诱导,表明它们是过渡状态。人类Mg_D细胞强烈表达先前用于描述“泡沫状”小胶质细胞的基因,例如脂质储存基因LPL和PPARG以及溶酶体基因CTSL,这支持了这些细胞在清除髓鞘碎片中的作用。人类Mg_E细胞与Mg_D细胞具有相似的特征,但额外表达NUPR1,这是一种与铁死亡相关的应激反应基因。人类Mg_F细胞在转录上具有独特性,其特征是VEGFA和FOSL2的高表达。最后,人类PVM细胞聚类的特征是巨噬细胞标志物(F13A1、MRC1、CR1和LYVE1)的特异性表达。总之,这些数据表明,与他们在小鼠脱髓鞘模型中观察到的相比,多发性硬化症病变的状态异质性更大。
使用Milo进行的DA检测表明,MS病灶中存在广泛的稳态偏离。虽然NAWM小胶质细胞表现出状态转变,这与先前报道的MS患者CNS中存在反应性小胶质细胞的结果一致,但与病灶组织相比,其变化幅度较小(图 6c)。除RLs外,所有病灶类型均显示稳态小胶质细胞显著减少(图 6c)。AL和CAL中DAM(Mg_D和Mg_E)显著富集,这与泡沫状小胶质细胞在髓鞘碎片丰富的区域聚集相一致。相反,CIL和RL优先富集DAM聚类Mg_F,而Mg_D和Mg_E的富集程度降低。Mg_F表达VEGFA,VEGFA可能促进血管生成并支持少突胶质细胞前体细胞(OPC)的存活和分化,提示该状态可能与修复相关。PVM 富集程度不高,主要局限于 AL 和 CAL。
通过DEG分析比较MS病灶与WM,发现小胶质细胞中存在广泛的转录重塑。NAWM的变化最少(共369个差异表达基因),而CAL的小胶质细胞变化最多(共955个差异表达基因)。所有病灶类型中仅有27个上调基因和20个下调基因是共有的,这突显了小胶质细胞的病灶类型特异性。AL和CAL共有32个上调基因和62个下调基因,而CIL和RL共有61个上调基因和17个下调基因,提示存在两条主要的激活路径,分别对应于炎症性病变和慢性/修复相关病变。在多种病变类型中,富集的通路包括UPR(BAG3、DNAJA4、TSPYL2)、mTORC1信号通路(HSPD1、HSPA4、SERPINH1)和上皮-间质转化(TGFBI、FERMT2、FLNA)(图 6d)。值得注意的是,CIL和RL小胶质细胞还富集了糖酵解(PKM、PYGL)和缺氧相关基因(VEGFA、PLIN2、FOSL2、NDRG1),这与LPC髓鞘再生DAM中观察到的代谢特征相一致。
为了直接比较小鼠和人类的小胶质细胞,他们进行了跨物种分析。MetaNeighbor 分析显示,细胞分为两个主要聚类,分别对应于稳态小胶质细胞和 DAM/PVM(补充图 15c)。在激活分支中,出现了两个亚聚类,一个包含小鼠和人类的 Mg_E/F 和 PVM,另一个包含小鼠和人类的 Mg_D 和循环小胶质细胞。小鼠 DAM 与 MS 富集的小胶质细胞共有40 个基因的保守上调,包括典型的 DAM 相关基因,例如APOE、LGALS1/3、VIM、CD44和CXCL16(图 6e),表明基本激活程序具有跨物种保守性。三重重叠特征显著富集脂质代谢基因(LPL、FABP5、PLIN2、ABCA1、COLEC12),反映了DAM在脱髓鞘后脂质处理中的核心作用。与CPZ DAM(17个独特的DEG)相比,LPC DAM与MS病变共有更多上调基因(脱髓鞘时55个独特的重叠DEG),表明LPC模型与MS炎症状态更为相似。LPC/MS特有基因富集溶酶体生物合成和脂质降解程序(GPNMB、NPC2、GALC、CTSZ、TPP1、IGF2R),与髓鞘碎片的主动吞噬作用一致;而CPZ特异性基因则提示慢性应激和代谢重编程反应(CRYAB、CPT1A、FGR、CDKN2B、GPX3)。各模型间下调基因重叠情况相似(LPC/CPZ/MS模型共有106个基因,LPC/MS模型特有58个基因,CPZ/MS模型特有48个基因),但值得注意的是,CAL模型的小胶质细胞差异表达基因最多,且与小鼠DAM下调模式最为相似(图 6e)。MS模型和两种脱髓鞘模型均表现出小胶质细胞稳态转录程序受到强烈抑制的特征,包括P2RY12、CX3CR1、ENTPD1(CD39)、BACH2、MEF2A和SALL1的下调。总之,这种跨物种比较揭示了MS模型和毒素脱髓鞘模型中保守的重塑过程,包括核心DAM特征基因的上调和小胶质细胞稳态特性的抑制。值得注意的是,LPC模型反映了溶酶体清除髓鞘碎片的能力增强,而CPZ模型则反映了慢性应激和代谢适应。
最后,基因特征分析进一步表明,缺氧和炎症相关程序在RLs中存在的人类Mg_F状态下最为富集,虽然炎症特征在人类Mg_D/E和PVM中也有轻微富集(图 6f)。相比之下,这两种特征广泛分布于小鼠DAM细胞群(Mg_C-E、PVM和增殖细胞)中,而非局限于某一亚型。总的来说,这些发现表明,毒素诱导的脱髓鞘模型捕捉到了一个保守的小胶质细胞激活核心,其特征是稳态特性丧失和共同炎症程序的诱导。然而,MS病变表现出更大的小胶质细胞异质性以及小鼠模型中未捕捉到的通路激活。这可能反映了人类疾病与急性毒素诱导的脱髓鞘相比,具有慢性、空间异质性的特点。或者,这可能表明小鼠小胶质细胞执行清除髓鞘碎片和创造有利于髓鞘再生的环境所需的多种功能,但这些功能在人类中可能多样化,并局限于特定的亚型,这与之前关于人类小胶质细胞进化分化的报道一致。
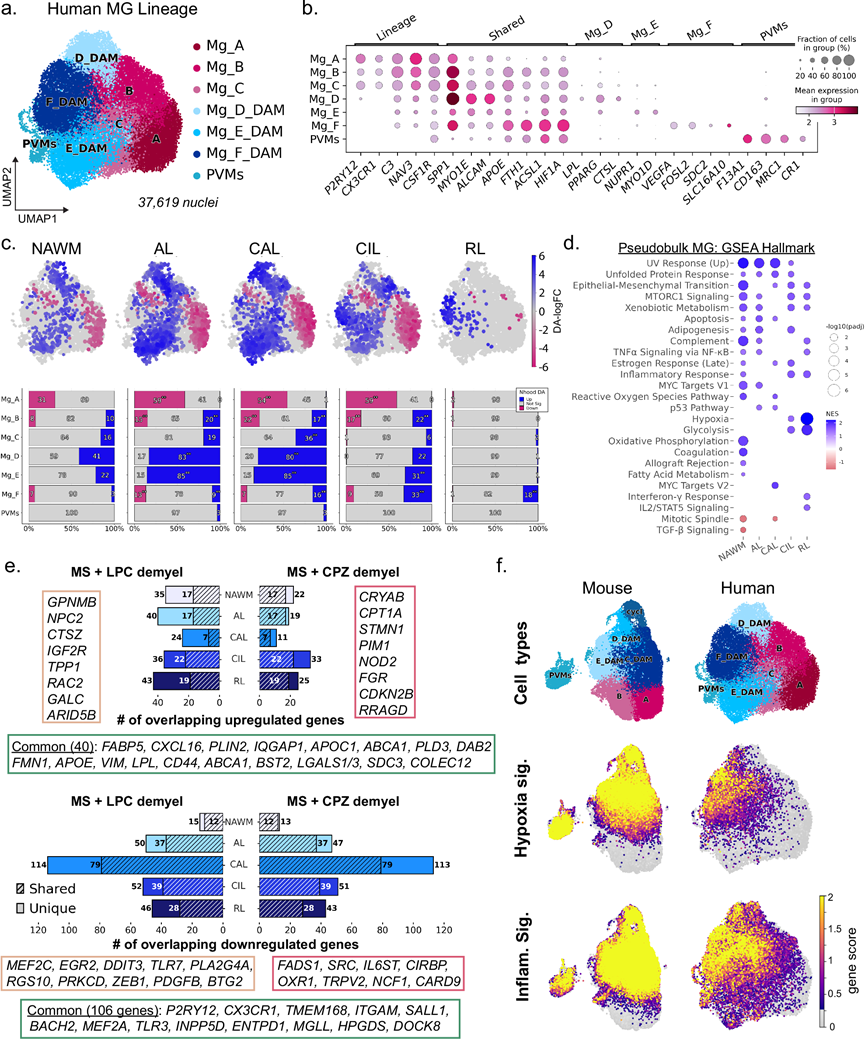
图6. MS 病变丰富了不同的小胶质细胞状态,这些状态与小鼠共有核心转录特征。
(a) 重新聚类的人类小胶质细胞/巨噬细胞谱系细胞的 UMAP 嵌入图。(b) 不同细胞类型中谱系和聚类特异性标记基因的散点图。(c) 差异表达细胞邻域叠加在 UMAP 嵌入图上(上图)。柱状图(下图)显示了与 NAWM 相比,按细胞类型注释的增加、减少或不变的邻域的比例。(d) 散点图显示了基于每种病变类型的假性大块 MG 的差异表达测试计算的 Hallmark 通路富集情况。(e) 条形图显示了脱髓鞘时 LPC 和 CPZ 中上调和下调的DEG与 MS 病变类型的重叠情况。(f) UMAP图显示了小鼠和人 MG 细胞,以及源自缺氧和炎症反应相关基因的基因评分。
+ + + + + + + + + + +
结 论
本文整合了来自CPZ和LPC诱导的脱髓鞘小鼠的新数据和已发表的多组学数据集,并将其与人类MS数据进行比较。本研究发现CPZ诱导了一种独特的、应激状态的少突胶质细胞(OL),其特征是Cdkn1a和Nupr1表达,类似于MS病变中的表型。在髓鞘再生过程中,这些模型均趋向于表达Socs3、B2m和干扰素反应基因的免疫反应性OL状态。小鼠小胶质细胞具有保守的激活程序,但LPC驱动的反应更强、持续时间更长。然而,这两种模型都未能捕捉到MS中观察到的少突胶质细胞祖细胞和小胶质细胞的异质性。这些结果提供了一个跨模型、跨物种的胶质细胞状态图谱,并为策略性地利用小鼠模型研究髓鞘损伤和修复提供了一个框架。
+ + + + +



