

 English
English文献解读|Nat Genet(29):从单细胞分辨率的多层组学数据中分析状态依赖性免疫特征
✦ +
+
论文ID
原名:Deciphering state-dependent immune features from multi-layer omics data at single-cell resolution
译名:从单细胞分辨率的多层组学数据中分析状态依赖性免疫特征
期刊:Nature Genetics
影响因子:29
发表时间:2025.07.28
DOI号:10.1038/s41588-025-02266-3
背 景
人体组学技术将生物学机制和疾病病理生理投射到具有不同分辨率的多层矩阵信息中。以种系遗传变异为基础的整合组学分析利用了分子数量性状基因座 (mQTL) 目录。多层 mQTL 目录协同分析了变异的功能注释,填补了从大规模人类疾病遗传学(即全基因组关联研究;GWAS)到结果临床表型的道路。此类努力最初从大量 RNA 表达谱开始,后来扩展到包括蛋白质组学和宏基因组学等高度多样化的层面。其中,单细胞转录组分析(scRNA-seq)的最新技术进展已成功阐明各种组织和环境中的细胞状态异质性。使用 scRNA- seq分析进行的遗传关联图谱可以捕获不同细胞类型中细胞状态下的连续遗传效应,并为人类性状相关遗传变异的分子机制提供更细致的了解。目前的单细胞表达 QTL(sc-eQTL)资源主要集中在欧洲血统,这使得在非欧洲血统中构建具有单细胞分辨率的多层组学成为可能。
实验设计
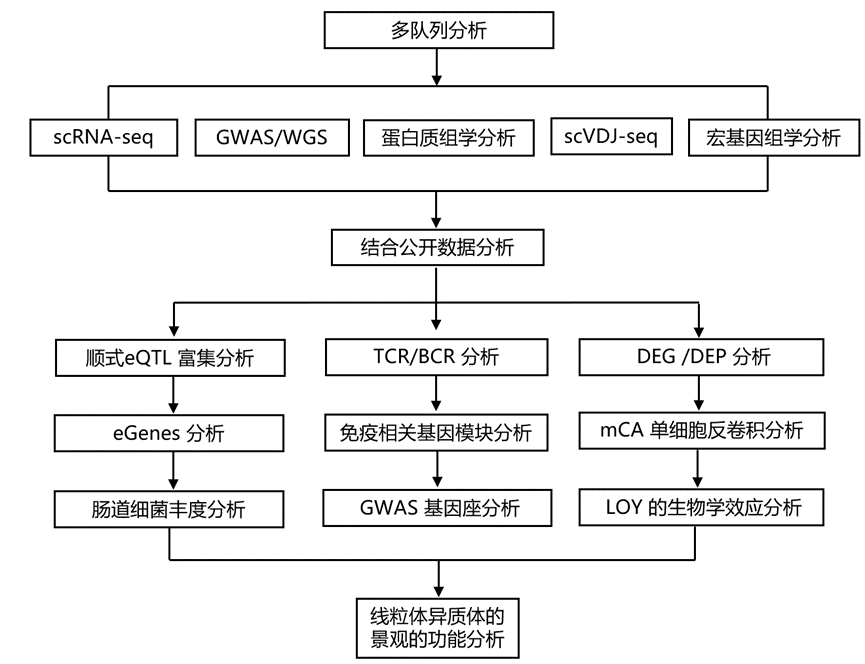
结 果
01

多层单细胞组学的大阪免疫细胞图谱(OASIS)队列分析
本研究纳入的OASIS 队列包括 88 名 COVID-19 患者和 147 名具有日本血统的健康个体(图1a)。研究团队使用 10x Genomics Chromium 平台对 2,059141 个外周血单核细胞 (PBMC)进行了scRNA-seq 和单细胞可变多样性连接(VDJ)测序(scVDJ-seq),获得了1506953个高质量细胞,根据已知标记基因的 RNA 表达手动注释细胞。他们首先根据 Azimuth L1 注释定义了七种主要细胞类型(L1)(图1b)。接下来,他们进一步鉴定了28种细胞状态(L2),并注释了10种细胞类型(LOneK1K)(图1b)。他们生成了用于表达数量性状位点(eQTL)分析和线粒体异质性检测的全基因组测序(WGS)数据,以及用于检测所有样本中嵌合性染色体变异(mCA)的单核苷酸多态性(SNP)阵列数据。他们使用对227个样本进行了血浆蛋白组分析,还对131份健康个体的粪便样本进行了鸟枪法宏基因组分析,并获得了系统发育相对丰度数据。
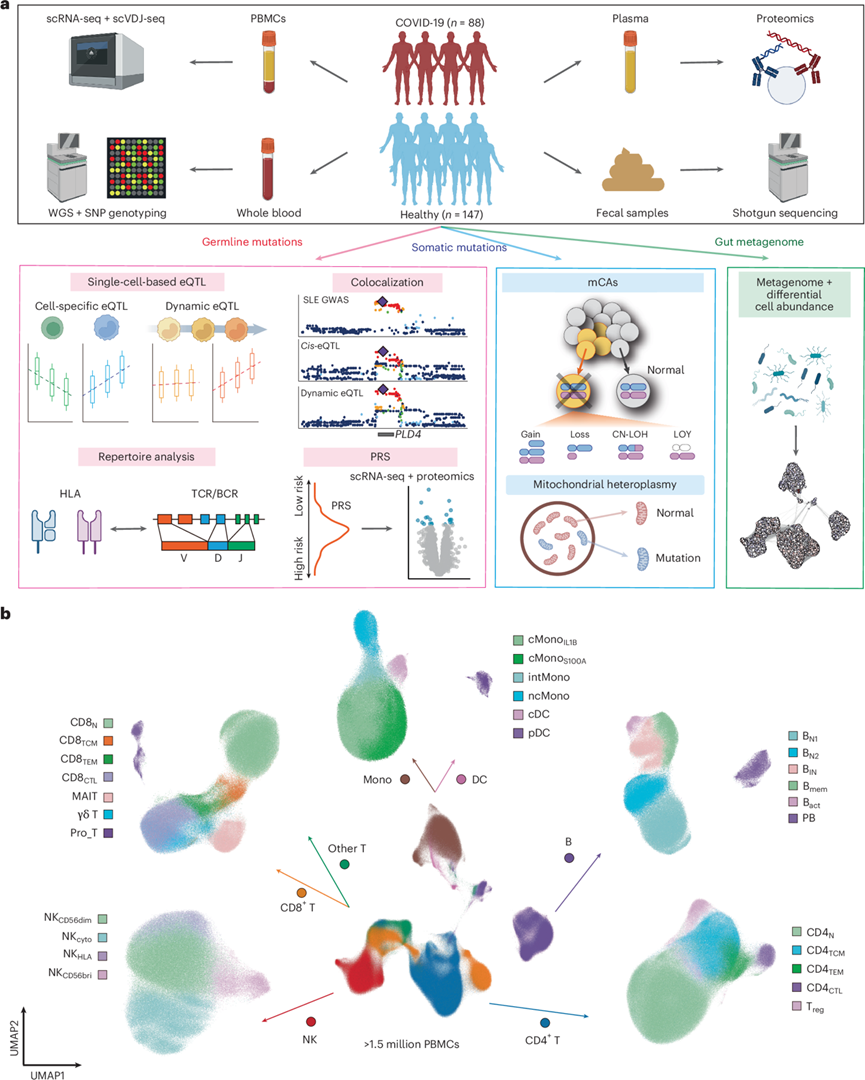
图1. OASIS 概览和 scRNA-seq 数据。
(a) 研究设计概述。(b) UMAP可视化。
02
免疫细胞类型特异性单细胞顺式-eQTL定位
为了评估免疫细胞中基因表达的遗传调控,他们首先使用伪批量方法,利用WGS数据进行单细胞顺式eQTL 分析,分别在 7 种主要细胞类型 (L1) 和 28 种细小细胞类型 (L2) 中,检测了位于转录起始位点 1 Mb 范围内的表达基因与遗传变异之间的关联,还绘制了条件独立的顺式eQTL图谱。
总的来说,他们在 L1 和 L2 中鉴定了 23443 和 34297 个 eQTL,其中 19641 和 30802 个为原发性顺式eQTL(图2a)。他们在 L1 中检测到 488-4901(中位数 = 3,176)个具有显著顺式eQTL效应的基因(eGenes),在 L2 中检测到 93-4062(中位数 = 862)个 eGenes,并且 eGene 的数量在不同细胞类型中差异很大。他们观察到 eGene 数量与相应细胞类型的每个样本的细胞数量之间存在很强的相关性。原发性显著 eQTL 效应大小与相应细胞类型每个样本的细胞数量呈负相关(图2b)。令人惊讶的是,该队列中每种细胞类型检测到的主要 eQTL 数量等于或大于 OneK1K的数量,而 OneK1K的样本量约为本研究队列的四倍。因此,他们通过在 L1 上进行下采样,广泛评估了顺式-eQTL 发现中样本量和每个样本的细胞计数之间的关系。当降低每个样本的细胞计数率以及样本量时,发现的 eQTL 数量呈线性下降,在 OneK1K 中也观察到了类似的关系。这些观察结果暗示检测 eQTL 的统计功效高度依赖于所分析的细胞数量。
接下来,他们评估了不同细胞类型之间 eGenes 的共有程度。他们观察到 8047 个 eGenes 中有 3422 个是细胞类型特异性的,而在 L1 中 1214 个 eGene 由五种以上的细胞类型共享(图2c)。在 L2 中,7386 个 eGenes 中有 2613 个仅在一种细胞类型中显著。他们还比较了不同细胞类型之间的 eQTL 效应,观察到 eQTL 共有水平很高,尤其在同一谱系内(T 和自然杀伤细胞、B 和髓系细胞)(图2d)。他们将显著 eQTL 的影响与来自日本个体的大量 eQTL 数据集进行了比较,发现一致性很高。
他们将本队列的顺式eQTL与欧洲人群OneK1K队列的顺式eQTL进行了比较,采用多变量自适应收缩方法。结果显示,OneK1K中显著的eQTL更易在OASIS对应细胞类型中复现,而OASIS中显著的eQTL在OneK1K中的复现率则较低,且在OASIS中eGenes数量多于OneK1K的细胞类型中,这一趋势更为明显。在初始CD4⁺ T细胞(CD4N)中,未在另一队列复现的eQTL其东亚与欧洲人群间次要等位基因频率(MAF)差异大于复现的eQTL。各细胞类型中显著eQTL效应的方向在另一队列中几乎完全一致。当收缩因子设为0.5时,十种细胞类型配对中,另一队列按效应量共有的eQTL中位比例分别为:OASIS eQTL 34.3%,OneK1K eQTL 69.3%。与复现情况类似,在两个队列间特有的CD4N细胞eQTL,其东亚与欧洲人群MAF差异也明显大于共有的eQTL。上述结果提示,基于更大样本量的不同人群构建单细胞eQTL资源具有重要意义。
他们利用路线图表观基因组学项目中八种免疫细胞类型的染色质状态预测注释了显著的 eQTL。主要 eQTL 在路线图免疫细胞的启动子和增强子区域均有富集。然而,启动子区域的 eQTL 富集在不同细胞类型中是一致的,而在增强子区域的 eQTL 富集则更具细胞类型特异性(图2e)。当按效应大小对主要 eQTL 进行分层时,启动子富集在 eQTL 的效应大小较大时更占优势,但无论效应大小如何,增强子的富集都相似(图2f)。考虑到自身免疫性疾病中超过 60% 的因果变异都映射到免疫细胞增强子,这一结果促使他们在增强子区域中鉴定更多不一定具有大效应大小的 eQTL。
为了在更精细的分辨率下揭示与细菌丰度存在差异的免疫细胞集,他们使用单细胞邻域(Milo)技术进行了差异丰度分析。在该分析中,他们重点关注以下三个物种,它们在比例分析中均与两种以上的细胞类型有显著关联,据报道与人类疾病有关:Ruminococcus gnavus、Prevotella copri和Bacteroides vulgatus。他们在 PBMC中鉴定了 43089 个邻域,其中没有一个与R. gnavus丰度显示出显著的差异丰度(图2g)。然而,R. gnavus的增加伴随着稀有细胞类型[即CD4+细胞毒性 T 细胞、活化 B 细胞和浆母细胞(PB)]的增加,以及髓系细胞类型的增加(图2h)。类似地,差异丰度分析显示,普氏菌(P. copri)的增加与浆细胞样树突状细胞和浆细胞样树突状细胞(PSC)的增加以及CD4 +细胞毒性T细胞的减少相关。此类差异丰度与比例分析的结果一致,表明了其稳健性。
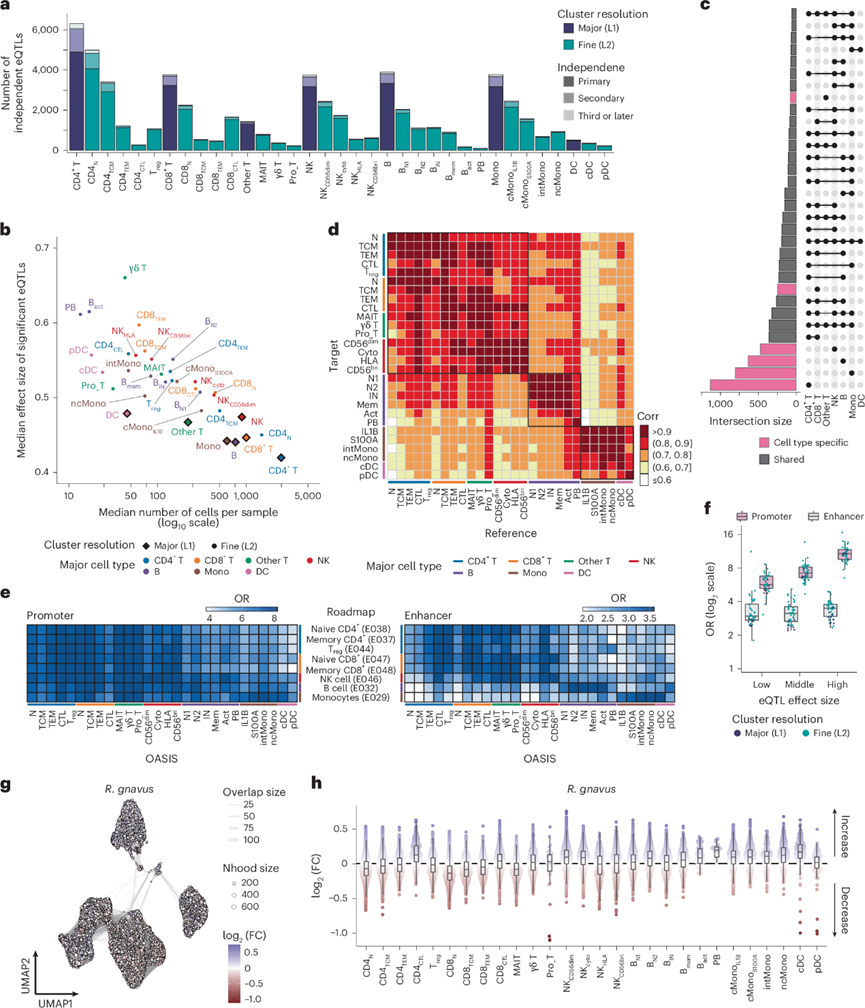
图2. 种系遗传变异和肠道宏基因组影响免疫细胞的转录和表型特征。
(a) L1 和 L2 级别每种细胞类型中显著的独立 eQTL 数量。(b) 散点图描绘了每个样本的中位细胞数与每种细胞类型中显著 eQTL 的中位效应大小之间的相关性。(c) L1级别细胞类型之间共有的显著 eGene 数量。(d) 热图显示 eQTL 效应大小的成对比较。(e) 热图描绘了来自路线图项目的八种代表性免疫细胞的启动子或增强子区域中 L2 细胞类型的主要显著 eQTL 的富集情况。(f) 主要 eQTL 的富集情况。(g) R. gnavus 相对丰度。(h) 蜂群图和箱线图显示了根据 L2 细胞类型中R. gnavus 的相对丰度。
03
HLA和全基因组与免疫系统的关联
他们进行了 scVDJ-seq 来构建 T 细胞受体(TCR)和 B细胞受体(BCR) 库的目录,包括每个基因在不同细胞类型中的使用情况。他们探讨了 HLA 与 TCR 库之间的关系,以特定细胞类型为特征。他们评估了 HLA 氨基酸变体与 TCR V 基因使用之间的关联,包括 TRAV 和 TRBV。他们发现在 HLA I 类和 II 类基因中,TRAV 基因与 HLA 氨基酸变体之间存在显著关联。具体而言,TRAV 基因分别仅与 CD8 + T 细胞和 CD4 N细胞中的 HLA I 类和 II 类基因变体相关(图3a-b)。这些结果与根据 T 细胞亚群的 HLA 对 TCR 的限制模式一致。此外,在CD4+ T细胞亚群之间的比较中,TRAV基因与HLA II类基因变异之间的关联在CD4N细胞中似乎比在整个(即CD4+T细胞)和中枢记忆CD4+ T细胞中更强(图3b)。这可能反映了在中枢水平(即胸腺中)形成的TCR库的HLA限制性比在外周水平形成的TCR库的HLA限制性更强。
他们探讨了HLA变异对COVID-19患者TCR库的影响,观察到显著的相互作用,尤其是HLA I类基因变异与CD8+T细胞中TRAV基因使用之间的相互作用(图3c)。这可能反映了SARS-CoV-2感染中CD8+ T细胞中HLA I类变异的不同,产生了不同的TCR库。II 类 HLA 位点与 CD4+ T 细胞中 TRAV 基因使用的主成分 (PC)2 以及 CD4N细胞中 TRAV 基因使用的主成分 (PC)1 之间存在显著关联(图3d)。
最后,他们对 BCR 进行了相同的组库特征范围关联分析,发现几种免疫球蛋白基因用法的 PC 与位于这些基因内的变异之间存在显著关联(图3e)。此外,K 链和 L 链的体细胞超突变 (SHM) 相关特征与 IGKV 和 IGLV 基因中的变异显著相关(图3e)。幼稚 B 细胞中更强的关联可以通过以下观察结果来解释:记忆细胞中的 SHM 更有可能受到暴露的影响,从而削弱遗传效应。
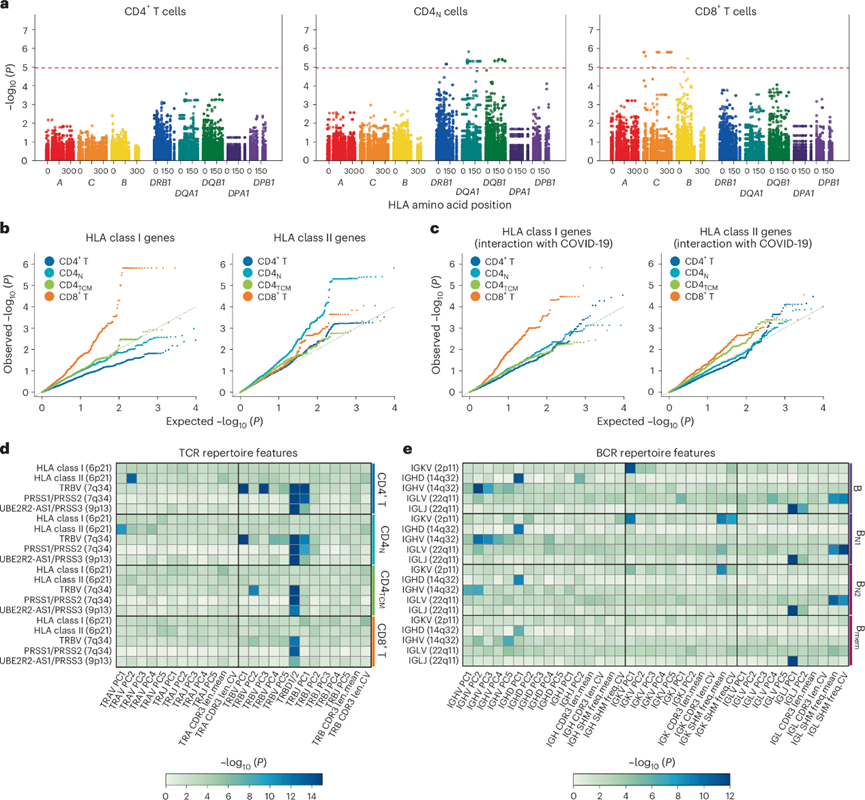
图3. HLA 和全基因组与 TCR 和 BCR 库的关联分析。
(a) 细胞中 HLA 与 TRAV 基因使用关联的区域图。(b-c) 不同细胞类型的HLA I 类(左)和 II 类(右)基因中 HLA 氨基酸变体与 TRAV 基因使用之间的关联和相互作用检验的分位数-分位数图。(d-e) 热图显示了 TCR和 BCR组谱中单个基因座(纵轴)内组谱特征关联。
04
两个免疫相关基因模块之间的动态 eQTL 效应
为了评估基因表达在连续细胞状态下的动态遗传调控,他们研究了与免疫和 COVID-19 严重程度相关的两个基因模块中髓系聚类中 eQTL 的动态效应(图4a)。模块 1与均匀流形近似和投影 (UMAP)1 高度相关,模块 2 与 UMAP2 高度相关,代表不同的细胞状态。为了对动态 eQTL 进行建模,将基因模块分成 10 个bin,并为每个模块重建每个bin中每个个体的平均表达谱(图4a)。他们分别使用线性和二次模型对两个模块测试基因型和分位数秩之间的相互作用来评估动态 eQTL,分别从稳健候选基因中鉴定出 530 和 568 个在模块 1 和 2 中具有动态 eQTL 效应的基因(deGenes)。在这些 deGenes 中,352 (66.4%) 和 393 (69.1%) 在线性和二次模型中均表现出动态 eQTL 效应,134 (25.3%) 和 117 (20.6%) 仅在模块 1 和 2 的二次模型中表现出动态 eQTL 效应。此外,超过一半的 deGenes(模块 1 中的 324 个和模块 2 中的 362 个)是模块特异性的(图4b),并且模块特异性 deGenes 的富集途径与模块 1 中的先天免疫和模块 2 中的抗原呈递有关(图4c)。
他们使用Roadmap数据对动态eQTL进行了注释,方法同顺式eQTL。动态eQTL在启动子和增强子中均表现出单核细胞特异性富集(图4d),但与顺式eQTL的富集模式明显不同(图2e)。随后,他们比较了动态eQTL与顺式eQTL在单核细胞(L1)启动子和增强子中的富集情况。在两个模块中,动态eQTL在增强子中的富集程度均高于顺式eQTL,而在启动子中则更低(图4e)。在髓系细胞亚群(L2)中(例如浆细胞样树突状细胞),动态eQTL与顺式eQTL的比较也观察到类似结果。他们进一步研究了那些动态eQTL位于功能区域、但在髓系细胞任一细胞类型中均不存在顺式eQTL的deGenes。对这些deGenes在各模块及功能类别中的通路富集分析显示,模块1富集了Toll样受体相关通路,模块2富集了抗原呈递相关通路;在每个模块内,其启动子与增强子中的富集模式亦存在差异(图4f)。
他们在单细胞分辨率下进行了动态eQTL分析(图4a)。以rs11080327对SLFN5的影响为例,他们观察到该位点仅在经典单核细胞(cMono)的特定状态下表现出强烈的细胞状态依赖性eQTL效应(图4g),其解析度高于既往报道。在15个hPC中,hPC14与I型干扰素通路相关,其细胞状态交互作用最为显著(图4g),与动态eQTL分析结果一致。类似地,NFKBIZ和IFITM2的eQTL(rs9818678、rs741738)也表现出显著的细胞状态依赖性,但其交互的细胞状态各不相同(图4g)。将eQTL定位拓展至单细胞分辨率,将为动态遗传调控提供更精细的图景。
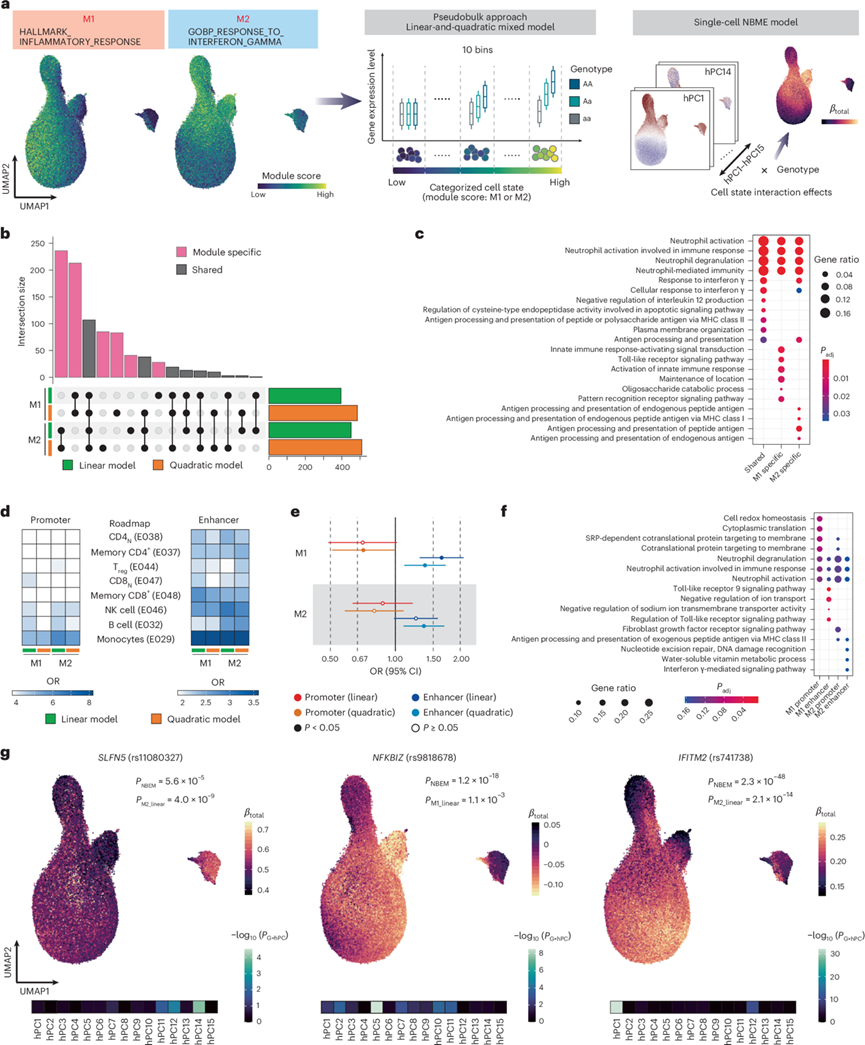
图4. 髓系聚类中的动态 eQTL 分析。
(a) 动态 eQTL 分析概述。(b) 两个模块的线性或二次混合模型中具有显著基因型-模块相互作用(即动态 eGenes)的 eGenes 数量。(c) GO分析。(d) 热图描绘了路线图项目中八个代表性免疫细胞的启动子或增强子区域中动态 eQTL 的富集情况,针对每种模块和分析模型的组合。(e) 森林图显示动态 eQTL与路线图中单核细胞功能区域重叠的 OR,与两个模块中功能区域和分析模型的每个组合中L1单核细胞中的顺式eQTL相比。(f) GO分析。(g) UMAP 表示每个细胞的细胞状态依赖性 eQTL 强度。
05
GWAS 变体和 sc-eQTL 的共定位
为了更好地理解 GWAS 基因座的遗传调控机制,他们评估了东亚人群中 13 种复杂性状的 GWAS 信号的共定位,并将eQTL 信号映射到 L1 和 L2 中每种细胞类型。他们在 121 个 GWAS 基因座上发现了 GWAS-eQTL 共定位事件,并优先考虑了 179 个候选性状相关基因(图5a)。这些 GWAS 基因座中约有一半(121 个中的 55 个)仅在一种主要细胞类型内显示共定位(图5a),并且大多数优先的性状相关基因是性状特异性的,表现出共定位的细胞类型明显特定于性状(图5a)。
接下来,他们将动态 eQTL 与 GWAS 基因座联合共定位。例如,rs2841281是PLD4基因座中系统性红斑狼疮 GWAS 的主要 SNP ,它仅与模块 1 有动态 eQTL 效应(图5b)。虽然这个变异与每种细胞类型的一些顺式 eQTL 中度共定位,但它与模块 1 的两个bin中的动态 eQTL 共定位更强(图5b-c)。此外,单细胞分辨率 eQTL 建模揭示了 IL1B 经典单核细胞(cMono IL1B)和 S100A 经典单核细胞之间的边界区域最强的细胞状态依赖性 eQTL 效应(图5d)。ETS2基因座中溃疡性结肠炎GWAS的领先SNP rs2836884,与模块1的动态eQTL效应比与模块2的动态eQTL效应更显著(图5e)。该变异与模块1第6个bin中的动态eQTL几乎完美共定位(图5e-f),并且即使在表现出显著共定位的细胞类型中,也表现出异质性细胞状态依赖的eQTL效应(图5g)。这些观察结果表明,在解释GWAS信号时,考虑动态eQTL的重要性。
除单变异外,他们以全基因组方式展示了动态基因调控。进一步探讨了多基因风险评分(PRS)如何在不同临床状态和/或细胞类型中影响转录组与蛋白组特征。使用PRS-CSx,结合欧洲人和日本人的住院COVID-19汇总统计作为训练数据,构建了住院COVID-19患者的PRS。该PRS在OASIS中解释的表型方差(R2)为4.1%,表明PRS具有合理的准确性。他们在COVID-19单核细胞和CD8⁺ T细胞中鉴定出差异表达基因(DEG),并在COVID-19蛋白质组中鉴定出差异表达蛋白(DEP)(n=184);而在健康个体中未检测到DEG或DEP(图5h,图S7a-b)。进一步确认,患者特异的DEG和DEP并不直接等同于患者与对照之间的DEG和DEP(图S7c-d)。他们还研究了来自其他性状GWAS的PRS效应,发现其作用具有情境和细胞类型特异性(图S7e)。这些结果表明,PRS与单个种系变异类似,可能在情境和/或细胞类型特异的方式下影响转录和蛋白表达谱。
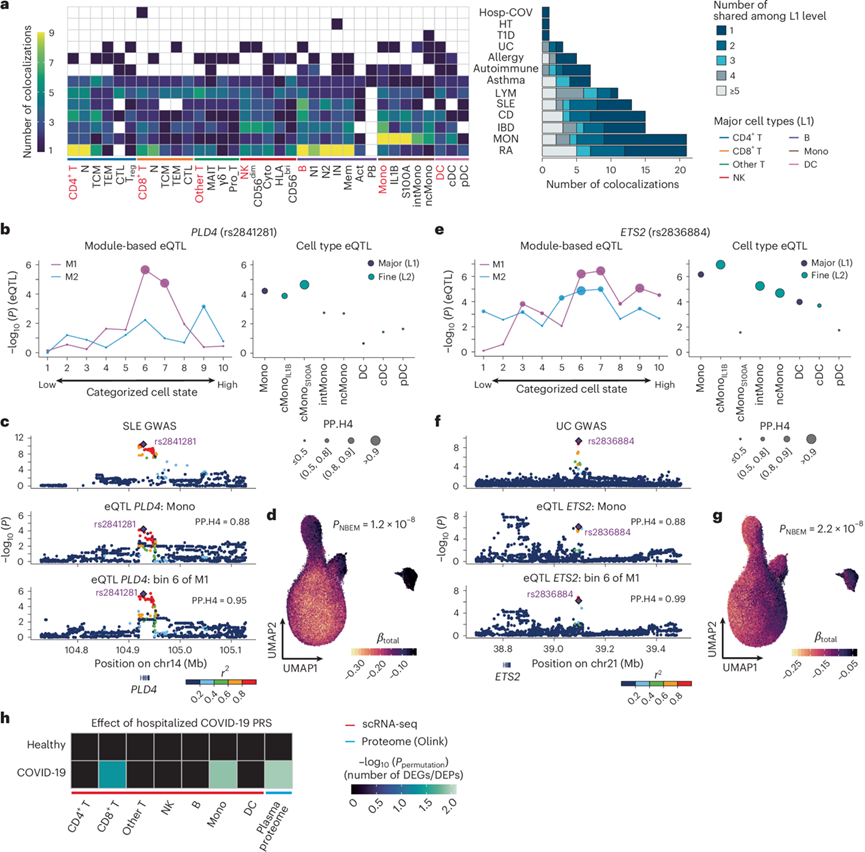
图5. 使用 OASIS 数据分析 GWAS 结果。
(a) 在每种细胞类型-性状组合中,与 eQTL 显著共定位的 GWAS 基因座数量(左)。主要细胞类型 (L1) 的细胞类型以红色表示。与 eQTL 共定位的 GWAS 基因座数量以及每种性状在主要细胞类型中共定位的次数(右)。(b) rs2841281细胞类型中PLD4的动态 eQTL 和顺式eQTL。(c) GWAS 的区域关联图,单核细胞的顺式eQTL (L1) 和模块 1 的 bin 6 中的动态 eQTL,针对PLD4基因座。(d) UMAP 表示每个细胞的细胞状态依赖性 eQTL 强度。(e)每个细胞类型的rs2836884的动态 eQTL 和顺式eQTL。(f) GWAS的区域关联图,(g) 单核细胞的顺式eQTL (L1) 和模块 1 的 bin 6 中的动态 eQTL,针对ETS2基因座。(h) COVID-19 PRS 对 COVID-19 患者和健康个体转录组和蛋白质组水平的影响。
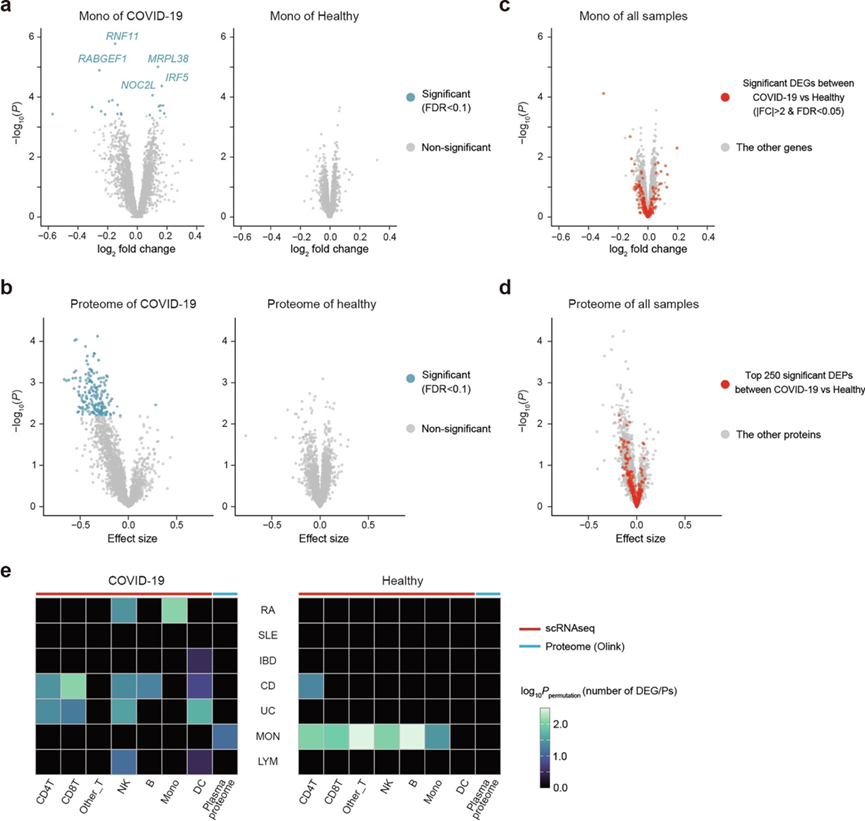
图S7. 多基因风险评分对转录组和蛋白质组的影响。
(a) 分别对COVID-19患者和健康受试者的单核细胞住院COVID-19多基因风险评分(PRS)进行差异表达基因(DEG分析。(b) 分别对COVID-19患者和健康受试者的血浆蛋白质组中住院COVID-19 PRS进行4分位数的差异表达蛋白(DEP)分析。(c) 所有样本的单核细胞住院COVID-19的DEG分析。 (d) 所有样本血浆蛋白质组中住院 COVID-19 PRS 的 DEP分析。(e) PRS 对 COVID-19 患者和健康受试者的 7 种自身免疫和血液相关性状的转录组学和蛋白质组学水平的影响。
06
mCA的单细胞分析
为了将mQTL 目录扩展到体细胞遗传变异,他们对用基因组学数据检测到的各种体细胞事件进行了单细胞反卷积分析(图6a)。使用 SNP 阵列数据,分别在 7个和 6 个样本中检测到 8 个拷贝数变异(CNA)和 6 个拷贝中性杂合性丢失(CN-LOH)。通过使用个体的 mCA 作为先验信息并将 Numbat 技术应用于 scRNA-seq 数据,可以区分每个样本中的突变细胞和野生型细胞,除了一名 COVID-19 患者含有较短的 CNA(CH12)。因为对具有 CN-LOH 的克隆细胞的检测依赖于 scRNA-seq 原始读数中嵌入的种系 SNP 等位基因,所以增加这种基于 scRNA-seq 的 SNP 信息可以提高灵敏度。因此,他们通过将目标深度从每个细胞 20000 扩展到 100000 个读数并将读取 2 长度从 90 扩展到 270 bp 进行了深度和长测序。因此,与正常测序条件相比,他们分析了 1.8 倍的 SNP 并鉴定出 2.1 倍以上的具有 CN-LOH 的突变细胞。虽然具有 CN-LOH 的细胞在其他 T 细胞中适度富集,但在两例 COVID-19 患者(CH01 和 CH05)中观察到具有 CNA 的细胞具有很强的细胞类型特异性(图6b)。这两名患者还显示出相对较大比例的突变细胞。
在 CH01 中,两个具有不同 CNA 的克隆在单核细胞中富集(图6b-c)。为了表征这些克隆,他们评估了单核细胞中突变细胞和正常细胞之间的 DEG。1p 丢失的单核细胞中大多数下调的基因和 15q 获得的单核细胞中上调的基因位于改变的染色体区域本身内(即顺式),突出了对突变细胞的准确检测。但有些显示跨染色体 DEG,包括 1p 丢失的单核细胞中 TNFAIP3 的上调,TNFAIP3是 COVID-19 特异性免疫反应的分子之一。他们进一步评估了突变体和正常克隆之间DEG的富集通路,发现免疫相关通路(如对细胞因子产生的正向调节)在1p丢失的突变体克隆中具有显著性(图6d)。
具有 17q 增益的突变细胞在 CH05 中的 B 细胞中高度富集(图6b-e)。突变体和正常 B 细胞之间的 DEG 通路富集分析表明,突变细胞中免疫相关通路上调,对类固醇激素的反应下调。这表明这些突变细胞可能会降低全身皮质类固醇治疗的有效性并导致更差的预后。此外,他们对 scVDJ-seq 数据的整合分析在 CH05 中发现了一个相当大的 BCR 克隆型,具有 17q 增益(图6f)。该 BCR 克隆型是所有样本中最大的克隆型,主要由幼稚 B 细胞组成,这与大多数由类别转换的 PB 组成的扩增克隆型不同。使用源自该扩增 BCR 克隆型的重组抗体,他们评估了它们对 SARS-CoV-2 主要抗原的反应性。有趣的是,BCR 克隆型不与任何测试的抗原发生反应(图6g),不支持通过对 SARS-CoV-2 感染的正常抗体反应进行克隆扩增。这些克隆扩增的突变 B 细胞可能降低了 SARS-CoV-2 感染中 BCR 库的多样性,这可能对该患者的抗体反应产生负面影响。他们在单细胞分辨率下对 mCA 的反卷积有可能阐明体细胞突变对免疫细胞功能和严重感染发展的影响。
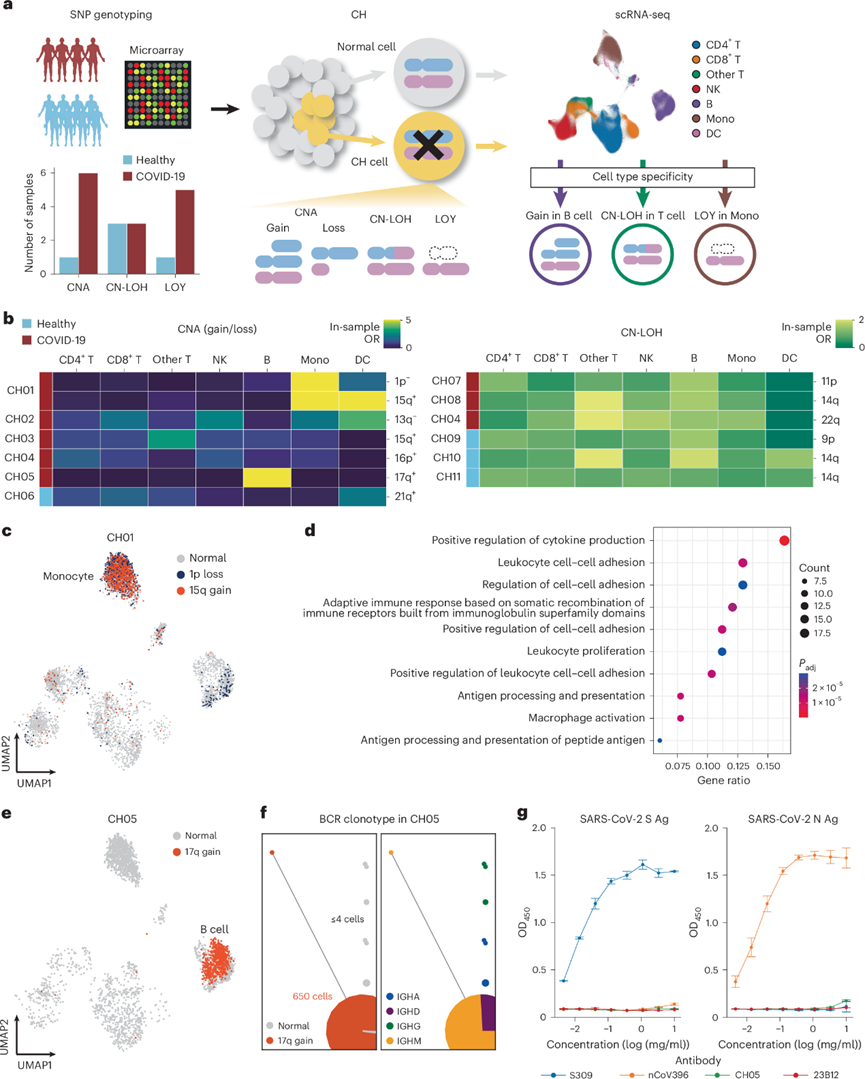
图6. mCA 的单细胞反卷积分析。
(a) 通过整合 SNP 阵列和 scRNA-seq 数据对包括 mCA 和Y染色体缺失(LOY)在内的 CH 进行单细胞反卷积的示意图。(b) 热图显示包含具有 CNA(左)和 CN-LOH(右)的细胞的每种细胞类型的样本内 OR。(c) UMAP可视化。(d) 通路富集分析。(e)UMAP可视化。(f) 网络图显示 CH05 的 BCR 重链和轻链中互补决定区 3(CDR3)氨基酸序列的相似性。(g) 酶联免疫吸附试验中针对 SARS-CoV-2 抗原 (Ag) 的抗体反应性。
07
单核细胞特异性Y染色体缺失(LOY)积累
接下来,他们评估了 LOY 对免疫系统的生物学效应。使用来自男性样本(n = 149)的 scRNA-seq 数据,将具有 LOY 的细胞定义为未从 Y 染色体的男性特异性区域表达的细胞,这使他们能够定量估计每个男性的 LOY 状态。年龄较大的男性显示出更大比例的 LOY 细胞。使用 SNP 阵列数据(即基于基因型的二元 LOY 估计)检测到的六个具有 LOY 的男性样本含有相对较多的 LOY 细胞(图7a)。因此,基于单细胞的方法对检测 LOY 更为敏感并提供生物学见解。对于基于单细胞的 LOY,所有男性样本均归类为高 LOY或低 LOY。利用此分类。虽然基于基因型的LOY没有显示出独立的影响,但基于单细胞的LOY与住院COVID-19风险显著相关(图7b)。
在 COVID-19 患者和健康个体(图7c-d)中,具有 LOY 的细胞在单核细胞中富集。具有 LOY 的 cMono IL1B细胞上调的 DEG 在 T 细胞相关通路中显著富集(图7e)。接下来,他们使用 Milo对 COVID-19 患者的LOY-high和 LOY-low样本进行了差异丰度分析。他们发现 LOY-high 样本中的单核细胞和树突状细胞增加,而幼稚 T 细胞减少(图7f),在健康个体中也观察到了类似的趋势。两组细胞比例比较表明,CD4+ T细胞中幼稚T细胞显著减少,而LOY-high的COVID-19患者调节性T细胞比例明显升高。总的来说,这些数据表明具有LOY的单核细胞可能通过T细胞成分的变化来影响免疫反应。
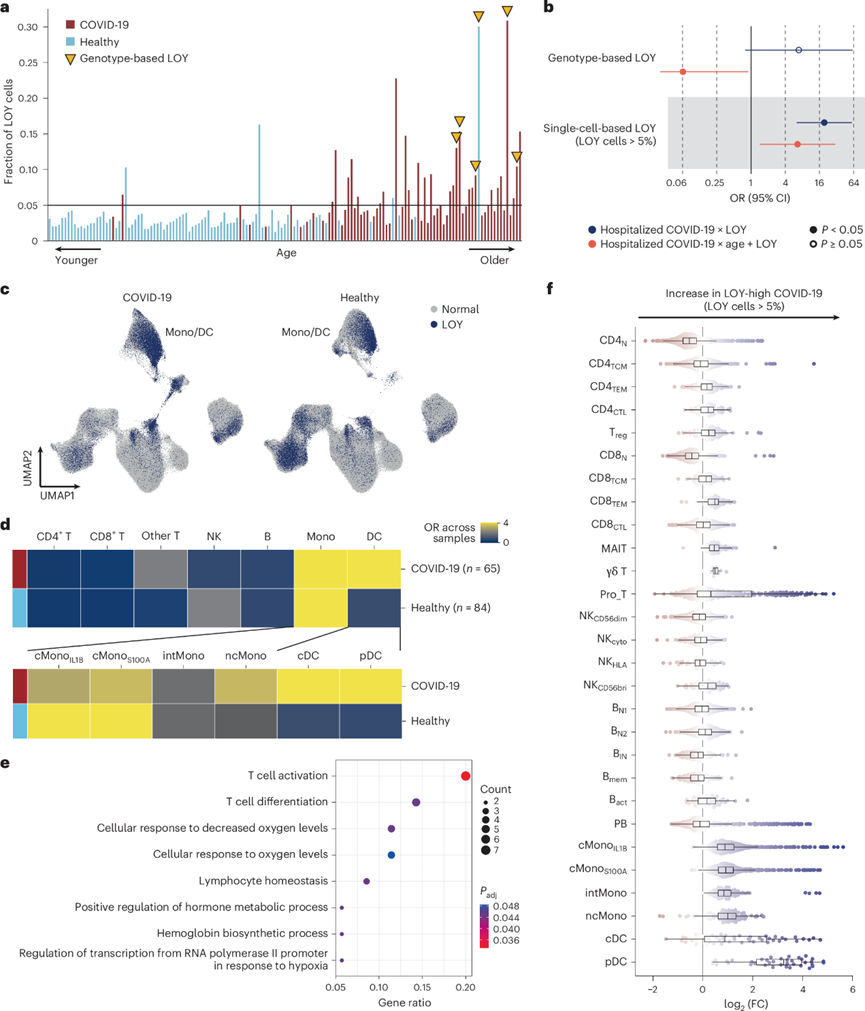
图7. LOY 的生物学效应。
(a) 按年龄(x轴)排序的每个男性样本中具有 LOY 的细胞分数。(b) LOY 与 COVID-19 风险之间关联的森林图。(c) UMAP可视化。(d) 热图显示 COVID-19 患者和健康个体中含有具有 LOY 的细胞的每种细胞类型样本的 OR。(e) 通路富集分析。(f) LOY-high 和 LOY-low 样本分布。
08
线粒体异质体的特异性富集
他们还通过整合基因组学和 scRNA-seq 数据研究了免疫细胞中线粒体异质体的状况(图8a)。基于基因组分析工具包 (GATK) 的流程使用 WGS 数据检测到 36 名(40.9%)COVID-19 患者和 63 名(42.9%)健康个体患有线粒体异质体(图8b)。他们成功地对 7 名 COVID-19 患者和 4 名健康个体的每个细胞进行了克隆分配。scRNA-seq 读段中具有线粒体异质体的细胞比例与 WGS 数据中线粒体异质体的 VAF 密切相关,m.813A>G 除外(图8c)。他们评估了这些样本中异质细胞的细胞类型特异性,但有一个健康样本除外,该样本中几乎所有细胞都具有线粒体异质体,具有线粒体异质体的细胞在单核细胞和树突状细胞中富集(图8d)。值得注意的是,m.813A>G 的异质体,其异质细胞分数与 VAF 之间的差异很大,表现出更强的细胞类型特异性。鉴于其位于 12S 核糖体 RNA 中,而 12S 核糖体 RNA 是与人类疾病有关的氨基糖苷类耳毒性的突变热点,这种异质体可能以细胞类型特异性的方式影响细胞增殖或活力,导致线粒体功能异常。
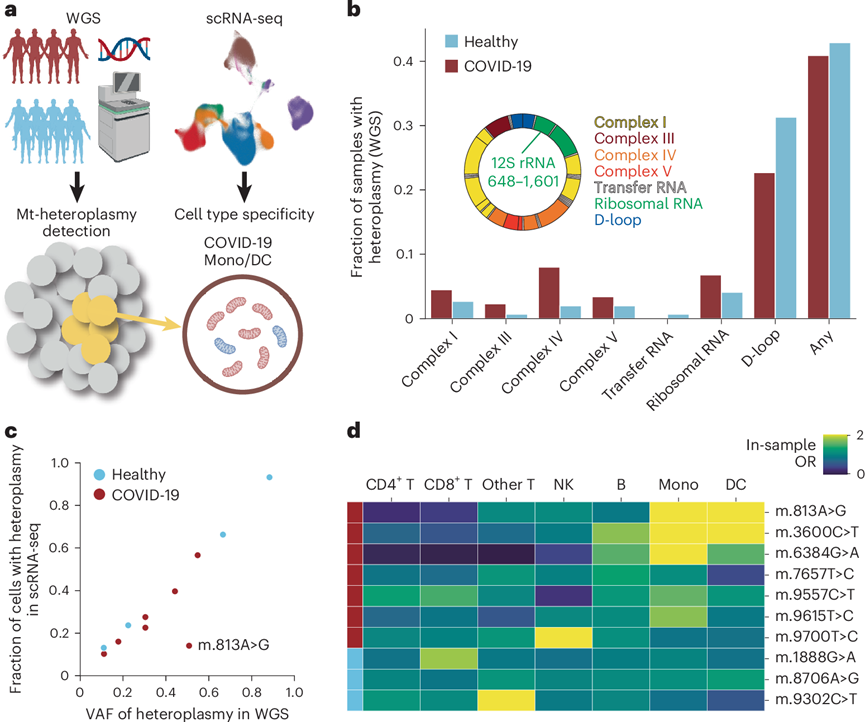
图8. 线粒体异质体的景观。
(a) 通过整合 WGS 和 scRNA-seq 数据对线粒体异质体进行单细胞反卷积的示意图。(b) 使用 WGS 数据按 mtDNA 区域检测的含有线粒体异质体的样本分数。(c) scRNA -seq 数据中含有异质体的细胞分数与 WGS 数据中异质体的 VAF 之间的相关性。(d) 热图显示包含含有线粒体异质体的细胞的每种细胞类型的样本内 OR。
+ + + + + + + + + + +
结 论
本研究构建了一个免疫细胞图谱,其中包含来自235名日本人(包括COVID-19 患者和健康个体)的超过150万个外周血单核细胞的单细胞转录组、宿主遗传学、血浆蛋白质组学和宏基因组学数据,分析了生殖细胞遗传学对不同免疫细胞类型和不同细胞状态下基因表达的影响,阐明了细胞类型和环境特异性的HLA以及与T细胞和B细胞受体库的全基因组关联。利用动态遗传调控进行共定位可以更好地理解全基因组关联信号。差异基因和蛋白质表达分析描绘了多基因风险对细胞类型和环境特异性的影响。各种体细胞突变,包括嵌合性染色体变异、Y染色体缺失和线粒体DNA (mtDNA)异质体,都投射到单细胞分辨率下。本研究鉴定了体细胞突变细胞特有的免疫特征。总体而言,免疫细胞以细胞状态依赖的方式进行动态调控,并具有多组学特征。
+ + + + +



