

 English
English文献解读|Nat Microbiol(20.5):宏基因组免疫球蛋白测序揭示健康人类肠道中微生物菌株的 IgA 包被
✦ +
+
论文ID
原名:Metagenomic immunoglobulin sequencing reveals IgA coating of microbial strains in the healthy human gut
译名:宏基因组免疫球蛋白测序揭示健康人类肠道中微生物菌株的 IgA 包被
期刊:Nature Microbiology
影响因子:20.5
发表时间:2025.01.02
DOI号:10.1038/s41564-024-01887-4
背 景
免疫球蛋白 A (IgA) 是从粘膜表面分泌的主要人体抗体。每天产生的 IgA 比所有其他抗体同种型的总和还要多,而且大部分分泌到肠腔中。在人体内所有分泌抗体的浆细胞中,估计有 60% 位于肠道中,以维持整个胃肠道中 IgA 的高水平并维持肠道稳态。IgA 缺乏的患者自身免疫性疾病、传染病的发病率增加,肠道细菌易位到肠道屏障的水平也增加,溃疡性结肠炎和克罗恩病等疾病与不同的IgA 结合能力和特异性有关。更好地了解健康和疾病状态下的 IgA 动态将促进先进疗法的开发,包括基于 IgA 的疗法,以精确调节肠道菌群。IgA-SEQ 是目前研究 IgA 结合的最先进技术,它结合细菌荧光激活细胞分选和 16S rRNA 基因扩增子测序,以研究体内与 IgA 结合或未结合的细菌。然而,由于该方法依赖于细菌 16S rRNA 基因扩增子测序(16S 测序),因此存在明显的局限性。
实验设计
结 果
01

宏基因组免疫球蛋白测序方法开发
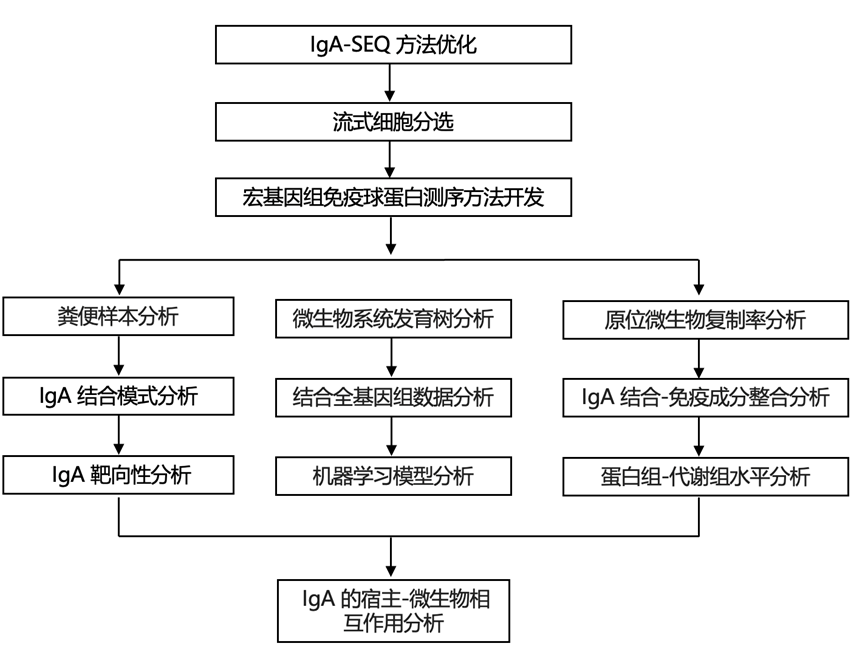
研究团队对 IgA-SEQ 方案进行了几项修改,以使其能够利用宏基因组测序,其中大多数修改旨在增加 IgA 结合的微生物细胞生物量(图1a)。为了通过荧光激活细胞分选 (FACS) 获得宏基因组测序所需的生物量,分选每个样本需要约 3 小时。因此,他们改用磁激活细胞分选 (MACS),这是一种更高通量和可并行的技术,还可以获得更高的生物量产量。在这里,他们使用 MACS 在 IgA+ 级分中实现了约 60% 的纯度(图1b-c)。通过比较原生和 IgA+ 级分中的微生物丰度水平,可以用这种纯度水平计算出 IgA 结合的准确测量值。为了确保使用该技术进行 IgA+ 细菌特异性富集,他们评估了细菌与对照磁珠的结合程度,发现几乎没有或完全没有 IgA 结合;然而,他们不能完全排除这代表来自具有 Fc 结合蛋白25的特定细菌的污染的可能性。最终的 IgA MIg-seq 方案在 IgA+ 微生物级分中产生了足够的生物质,从而能够进行深度宏基因组测序(IgA+ 级分的平均提取 DNA = 5.1 ± 0.6 ng)。
接下来,他们将 IgA结合的宏基因组免疫球蛋白测序 (MIg-seq)方法(IgA MIg-seq) 应用于一组在饮食干预过程中收集的成人粪便样本。他们对 38 个粪便样本进行了 IgA MIg-seq,从而获得了 76 个宏基因组(每个样本一个原生部分和一个 IgA+ 部分)[平均测序深度 = 16.2 ± 0.4 千兆碱基对 (Gbp)]。38 个粪便样本中有 5 个(13%)未通过流式细胞分析质量控制 (QC),其余 33 个粪便样本中有 3 个(9%)的原生或 IgA+ 部分低于最低测序深度 12 Gbp。因此,他们对通过 QC 且具有足够测序深度的 30 个粪便样本进行了分析。这 30 个样本来自 19 位不同的参与者,其中对11 位参与者采集了 2 个样本(一个在饮食干预之前,一个在饮食干预结束时)。本研究中的原生宏基因组与 IgA+ 样本一起进行处理,确保两个样本之间的所有差异都是 IgA 分类的结果,而不是技术差异。
在本研究分析的 30 个样本中观察到相同的一般 IgA 结合模式。变形菌在 IgA+ 级分中明显更丰富,而拟杆菌在 IgA+ 级分中明显减少(图1d)。IgA+ 级分的Shannon多样性低于原生样品(图1e),这是意料之中的,因为 IgA+ 微生物组是原生微生物组的一个子集。IgA+ 和原生级分分别显著富集了 27 种和 34 种细菌(图1f)。Bariatricus comes和Alistipes shahii分别是 IgA+ 和原生级分中最富集的物种。虽然样本是作为饮食干预的一部分收集的,但饮食干预对 IgA 结合水平或原生或 IgA+ 部分中的微生物组成没有显著影响。 同一个体的纵向样本之间的微生物组成和 IgA 结合微生物的总体比例随时间的变化类似。 最后,IgA+ 部分在参与者之间的差异明显大于原生样本,表明 IgA 靶向性在个体之间比整体微生物组成更具差异性。
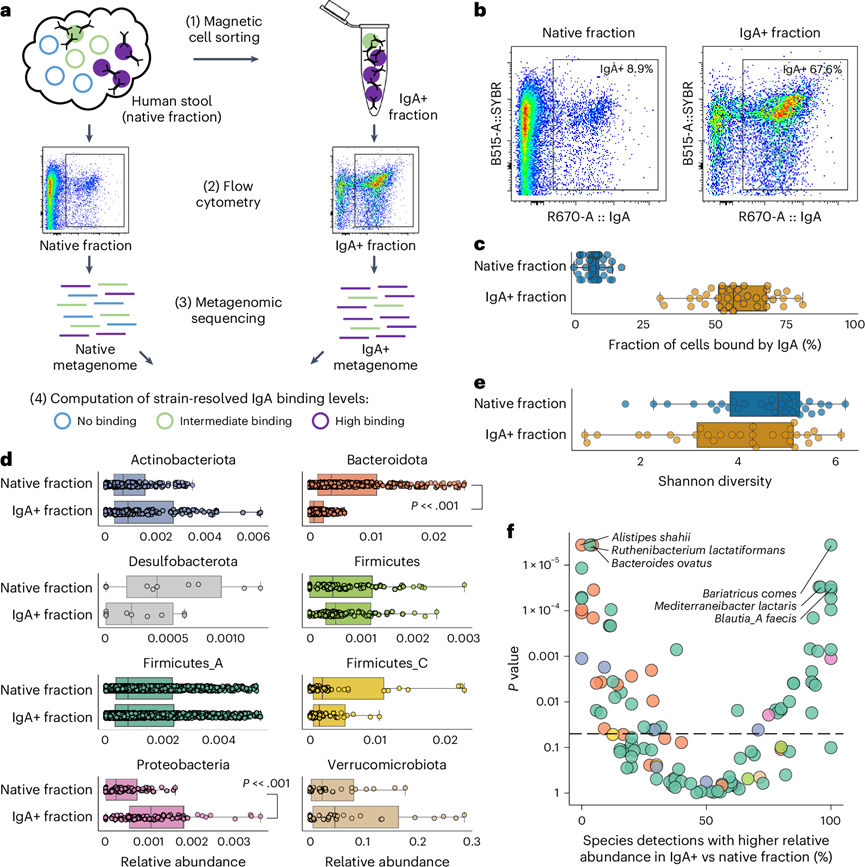
图1. 宏基因组免疫球蛋白测序 (MIg-seq) 是一种有用且强大的技术。
(a) 基于 IgA 的 MIg-seq 工作流程示意图。(b) 通过磁性细胞分离分类的原生和 IgA+ 级分的代表性流式细胞分析图。(c) 通过流式细胞技术检测的原生样品和 MACS 分类的 IgA+ 级分中与 IgA 结合的总微生物细胞百分比。(d) 按门分组的原生级分与 IgA+ 级分中物种的相对丰度。 (e) 原生(上)和 IgA+(下)宏基因组的 Shannon 多样性。(f) 对于每种微生物物种(点),该物种在 IgA+ 与原生部分中具有更高相对丰度的时间百分比(x轴)与关联的P值。
02
健康人体内 IgA 结合值一般是一致的
接下来,他们使用最近开发的 IgA+ 概率得分指标分析了单个菌株的 IgA 包被水平,该指标是根据 IgA+ 与原生部分中分类单元的相对丰度比率计算得出的。当对本研究中计算的所有菌株水平 IgA+ 概率得分进行排序分布时,出现了三个不同的划分(图2a)。他们将这些组归类为“无包被”(IgA+ 概率 < 0.005;21% 的物种检测)、“低结合”(0.005 < IgA+ 概率 < 1;61% 的物种检测)和“高结合”(IgA+ 概率 > 1;18% 的物种检测)。变形菌门和厚壁菌门物种在与 IgA 结合高的物种中显著富集,而拟杆菌门在无包被物种中富集(图2b)。他们鉴定出 20 个属,其 IgA+ 概率得分显著高于其他分类群,其中许多属于颤螺菌科(一个包含几种与人类健康呈正相关的物种的分类科)。因此,他们检验了 IgA 可能不成比例地覆盖与健康相关的菌株的假设。为了探究这一点,他们将四种确定为改善人类健康的活生物治疗主要候选菌属的微生物(双歧杆菌、粪杆菌、阿克曼氏菌和真杆菌)归为“益生菌”。值得注意的是,在本研究的样本中未检测到乳酸杆菌,这限制了将研究结果与早期报告健康参与者中IgA 与乳酸杆菌结合的研究进行比较的能。他们发现这些益生菌比其他类群更频繁地与 IgA 结合(图2c)。这一发现与之前的报告一致,可以用以下方式解释:益生菌(1)在肠道中占据一个具有更高宿主-微生物免疫相互作用水平的生态位,(2)受 IgA 促进和(3)驱动 IgA 的产生。
为了在物种水平上检验 IgA 结合情况,他们生成了在至少 5 个样本中检测到的所有微生物的系统发育树,并叠加了 IgA 结合数据(图2d)。在本研究中,物种水平的 IgA 结合模式在个体和样本之间非常一致,表明人类 IgA 对微生物的靶向性总体上是保守的(图2d)。虽然观察到了与前面提到的系统发育保守性一致的明显趋势(例如拟杆菌门菌株通常未包被,而变形菌门菌株通常包被较多),但他们还发现某些菌株表现出独特的结合模式,与它们的系统发育组不同。例如,脆弱拟杆菌即使属于整体 IgA 结合水平最低的门(拟杆菌门),也表现出高水平的 IgA 结合(图2d)。检测到的 IgA 涂层模式与其系统发育不同,这表明菌株内具有驱动 IgA 结合的独特遗传内容。
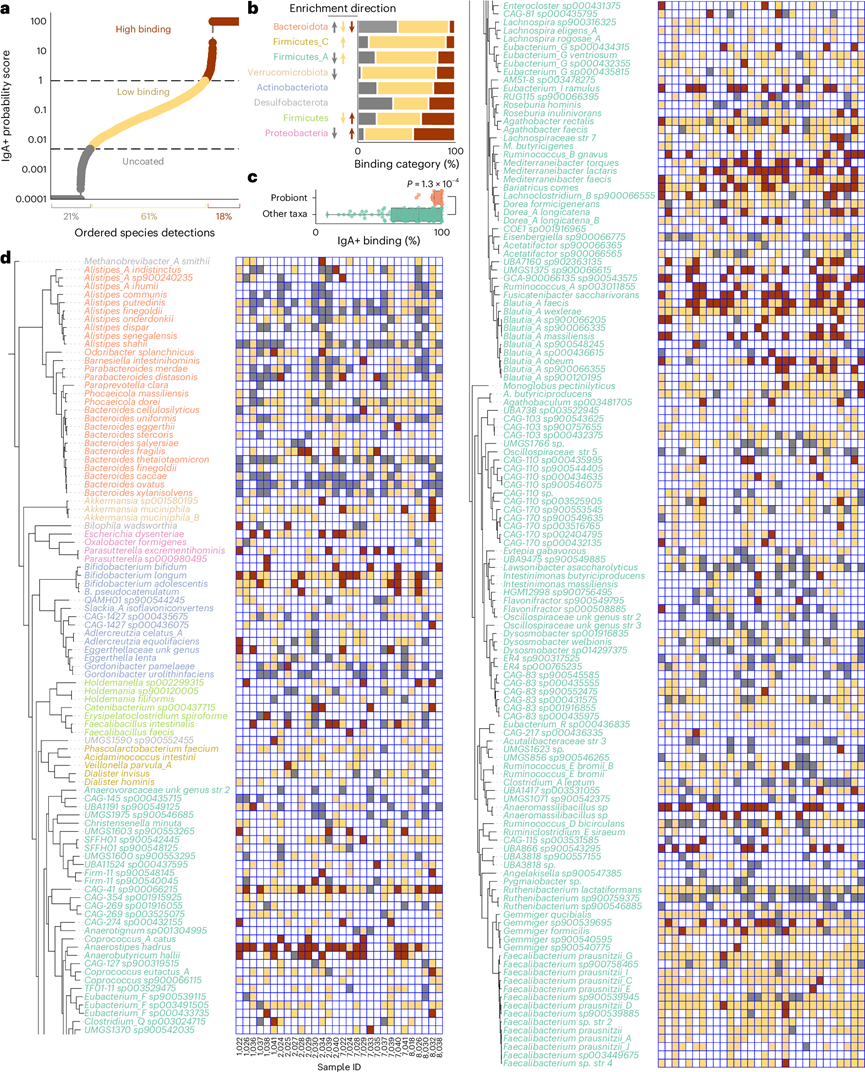
图2. 健康人体内的 IgA 结合水平一致。
(a) 所有样本中检测到的所有物种的对数尺度 IgA+ 概率分数,按等级排序。(b) 每个门内显示不同结合水平的检测百分比。(c) 益生菌与其他属中 IgA 结合率高或低的检测百分比。(d) 在至少 5 个样本中检测到的所有物种的连锁基因系统发育树。
03
特定微生物基因与 IgA 结合增加有关
接下来,他们利用 IgA MIg-seq 提供的全基因组测序数据来研究微生物基因与 IgA 结合之间的联系。经过训练的随机森林机器学习分类器根据微生物基因注释预测微生物 IgA 结合水平,总体准确率为 62%(图3a)。该模型在分类“无包被”和“高结合”物种方面比分类“低结合”物种更准确[曲线下面积 (AUC) 分别为 0.81、0.88 和 0.69],因为“高结合”和“无包被”微生物经错误归类为“低结合”的次数比彼此的次数多(图3b)。这些数据突出了微生物基因含量与 IgA 结合水平之间的紧密关联以及结合水平的层次性质(例如:高结合微生物的基因含量与低结合微生物的相似性高于与无包被微生物的基因含量)。
为了确定微生物基因含量与分类学与 IgA 结合的相对重要性,他们根据(1)分类学、(2)基因含量或(3)分类学和基因内容训练了额外的机器学习分类器。基于基因含量的分类器比基于分类学的分类器更准确,但同时可以访问分类学和基因内容的分类器表现并不比仅可以访问基因内容的分类器更好(图3c)。从这些发现中得出两个假设:(1)某些基因独立于分类学调节微生物的 IgA 结合;(2)一些与 IgA 结合相关的基因表现出很强的分类学联系。
考虑本研究中所有样本中检测到的所有微生物时,糖苷水解酶家族 101(GH101)成为与高 IgA 结合最显著相关的注释(图3d-e)。其次是 otrC(一种多药耐药外排泵)和 CAT RNA 结合结构域(一种与调节糖代谢操纵子相关的 Pfam 结构域)。GH101 家族成员是内切糖苷水解酶,通过从宿主粘蛋白中释放O-聚糖参与粘液降解。他们在 3 个门和 8 个属的 10 个基因组中鉴定了 GH101,包括长双歧杆菌、地中海杆菌和Odoribacter sp900544025种。该基因的广泛分类分布、与 IgA 结合的密切关联以及与宿主粘液降解相关的已知功能,支持存在增加 IgA 结合的微生物功能,无论分类如何。此外,这些结果表明,与预测作用于植物底物的 CAZymes 相比,参与粘液降解的 CAZymes 通常与高 IgA 结合更相关(图3f)。
在 Firmicutes_A 中还鉴定出一组 10 个与 IgA 强显著关联的注释(图3g)。与 GH101 类似,它们的功能主要涉及与粘蛋白和粘液成分的分解或相互作用相关的酶和结构域。有趣的是,检查其他门类中 IgA 涂层与富集功能之间的关系时,会呈现出不同的绘图结构,可能表明门级 IgA 关联不同。例如,变形菌显示的与 IgA 结合负相关的注释远多于正相关的注释(图3g),这可能表明 IgA 限制了该门的定植,并且某些基因已经适应以逃避结合。在所有属中,拟杆菌属具有最多与高 IgA 结合显著相关的注释(图3h)。由于脆弱拟杆菌属表现出比其他拟杆菌属高得多的 IgA 结合(图2d ),他们接下来研究是否特别是这个物种驱动了检测到的高 IgA 注释。与拟杆菌属中的 IgA 结合最正相关的基因,包括N -乙酰神经氨酸差向异构酶(一种参与粘液衍生碳水化合物代谢的基因),在脆弱拟杆菌中非常普遍(图3h),这一发现证明了与 IgA 结合和分类学密切相关的基因的存在。
图3. 分类单元特异性微生物基因与高 IgA 靶向性相关。
(a) ROC 曲线显示随机森林分类器根据基因内容预测 IgA 结合的性能。(b) 混淆矩阵显示真实与预测的 IgA 结合分类。(c) 使用双侧 Wilcoxon 秩和检验对分类法、基因内容或两者进行训练的随机森林分类器的准确度比较。(d) 对于每一种微生物功能(点),该功能与 IgA 结合的关联。(e) 左:条形图比较编码 GH101 的微生物中高 IgA 与低 IgA 或无 IgA 涂层的百分比。右:箱线图比较编码或不编码 GH101 的微生物的 IgA + 概率得分。(f) CAZyme(点)与 IgA 结合的关联。(g) 对于每种微生物功能(点),该功能与 IgA+ 概率得分的关联(x轴;编码功能的微生物的 IgA+ 概率得分中位数-不编码功能的微生物的 IgA+ 概率得分中位数)与功能与 IgA+ 概率得分的关联的P值。(h)与(d)相同,但仅考虑拟杆菌属的物种。
04
与 IgA 结合的细菌原位复制率较低
接下来,他们使用由 IgA MIg-seq 生成的宏基因组测序数据,通过计算本研究中检测到的所有微生物的原位微生物复制率(以 iRep 值衡量),来评估细菌复制率与 IgA 结合之间的关系。iRep 值受菌株特异性特性(如 DNA 聚合酶速度和基因组长度)的影响;他们通过仅比较在同一粪便样本的 IgA+ 与原生部分中检测到的同一菌株的值来控制这一点。在具有低或高 IgA 涂层的物种中,原生部分的复制率明显高于 IgA+ 部分(图4)。在不同微生物门和属中,复制率降低的幅度也存在显著差异,其中拟杆菌门在 IgA+ 样品中 iRep 的下降幅度大于厚壁菌门和厚壁菌门_A,这些数据支持 IgA 能够影响细菌复制率,并强调了 IgA 结合在不同分类群中的影响不同。这些数据还支持“链式生长”模型,该模型提出多反应性 IgA 可能会选择具有高复制率的细菌。其他解释包括:(1) 基于 IgA 的细菌聚集可能会限制聚集体中心细菌的营养获取和复制速度,和/或 (2) 持续 IgA 包被的微生物的代谢策略可能与生长率改变有关。
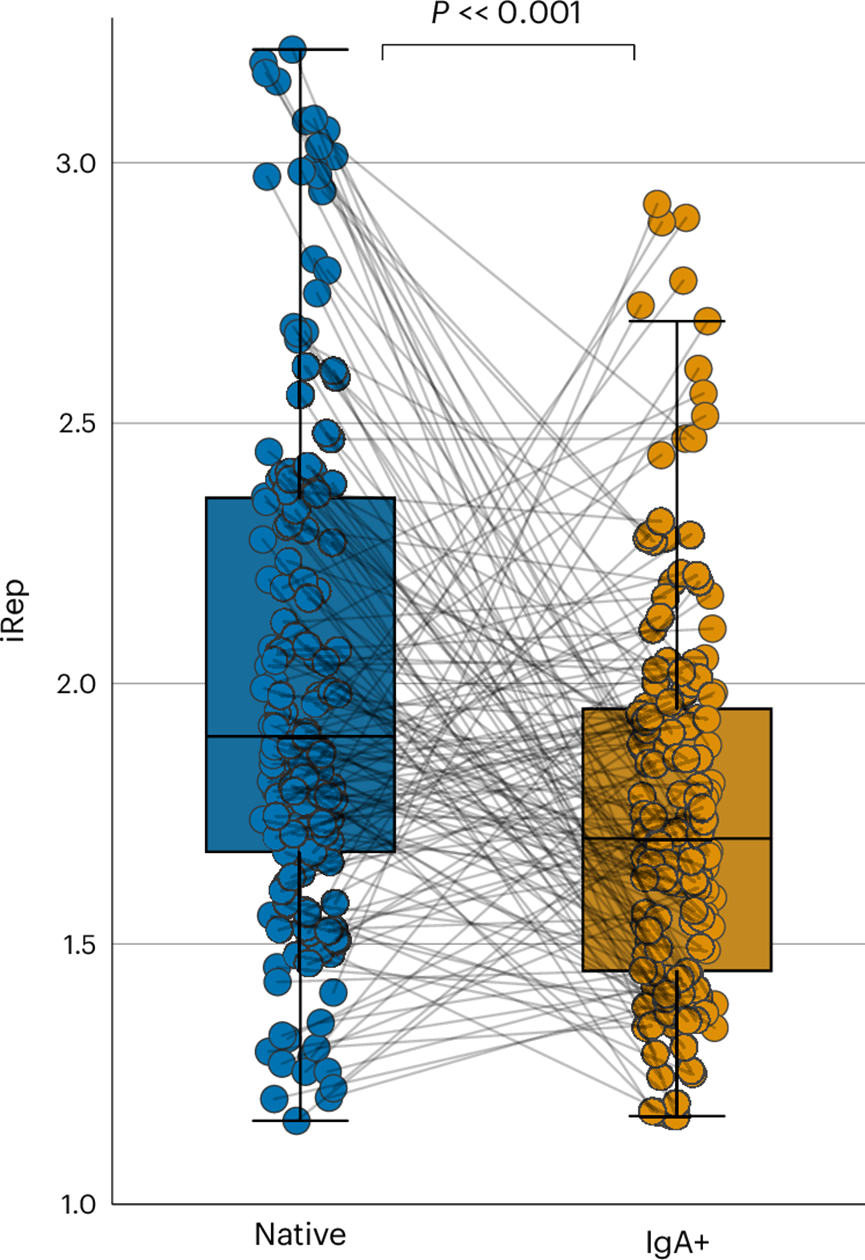
图4. IgA 结合影响原位微生物复制率。
同一粪便样本中原生(左)和 IgA+(右)部分中 IgA 结合率低或高的物种的复制率 (iRep) 的配对比较。
05
IgA 涂层与其他免疫成分密切相关
他们观察到免疫和健康数据与原生和 IgA+ 相对丰度值以及门和物种水平的许多相关性(图5a-b)。然而,IgA+ 概率得分(IgA+ 与原生相对丰度值之比)与免疫和健康数据显示出更强的相关性(图5a-b)。对 IgA 涂层和原生相对丰度的综合评估增强了信息价值,并表明 IgA 结合是微生物组-免疫系统相互作用背景下高度相关的测量指标。
从门水平分析与宿主数据发现了许多明显的关联(图5c)。甘油三酯水平与 Firmicutes_A 和 Actinobacteriota 的 IgA+ 结合有关,表明与 IgA 靶向存在潜在的代谢联系。Firmicutes_A IgA 结合也与中枢记忆 CD8 T 细胞高度相关。拟杆菌IgA 结合与纤维摄入量呈负相关,支持饮食在塑造针对特定微生物的 IgA 反应中的作用。乳酸水平与放线菌门的 IgA 结合相关,放线菌门是一门含有明确描述的乳酸生产菌,强调了宿主的免疫反应与肠道代谢特征之间的相互作用。与其他短链脂肪酸类似,乳酸可以由免疫细胞感知。
三种不同的粪杆菌(一种通常与炎症性疾病呈负相关的细菌属)的 IgA+ 概率评分与翻译延伸因子 G、细胞因子 CCL4 和细胞因子 TRANCE 的水平显著相关(图5d)。由于进行的比较次数较多且每个物种的数据点数量较少(与门级别相比),因此在物种水平上发现的显著相关性可能较少。IgA 相关基因与短链脂肪酸或饮食因素之间没有关联。总的来说,这些结果说明了 IgA 靶向如何与人类的生理和健康状况相结合,并且这种检测 MIG-seq 是揭示该集成系统机制的有力工具。
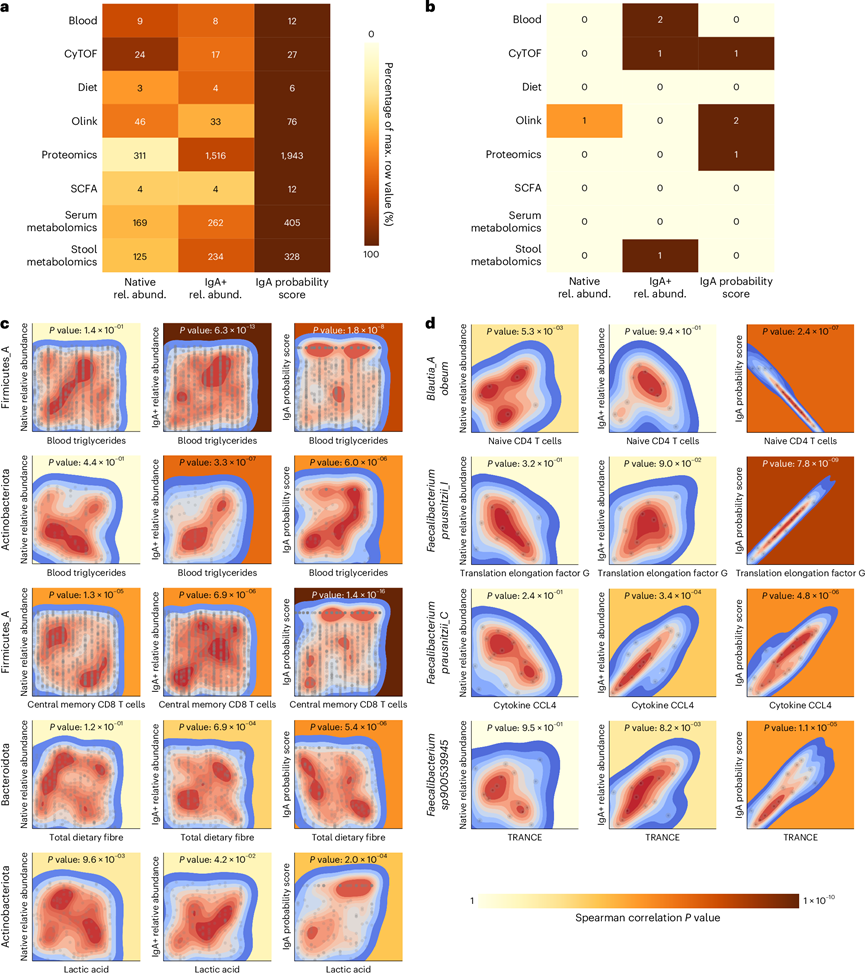
图5. IgA 结合与免疫系统的其他成分整合。
(a-b) 3 种不同的微生物测量值(x轴)与 8 种人类健康和微生物组功能测量值(y轴)之间的门级和物种级相关性分析。(c-d) 选定门和物种以及宿主衍生数据的本地微生物相对丰度(左)、IgA+ 相对丰度(中)和 IgA+ 概率得分(右)的相关性、密度和散点图矩阵。
+ + + + + + + + + + +
结 论
本研究开发了一种技术,即宏基因组免疫球蛋白测序 (MIg-seq),该技术可提供由 IgA 包裹的微生物的菌株水平分辨率,并使用它来确定健康人类粪便中 3520 种肠道微生物组菌株的 IgA 包裹水平。本研究发现健康和疾病相关细菌都是 IgA 的靶标。微生物基因对 IgA 结合水平具有高度预测性;特别是,粘液降解基因与高结合相关,并且与 IgA 结合的微生物的复制率显著降低。与传统的微生物群落组成相对丰度测量相比,IgA 结合与宿主免疫状态的相关性更高。这项研究介绍了一种评估人类粪便中菌株水平 IgA 结合的强大技术,为更深入地了解基于 IgA 的宿主-微生物相互作用奠定基础。
+ + + + +



